Basis sets
Vibrational basis set
TROVE provides a number of basis set options for the vibrational basis functions. Currently, all these options are 1D basis sets. With an exception for the Harmonic and Morse bases, all basis set are generated numerically by solving some model 1D Schrödinger equations. The solution methods can vary but are typically based either on the Numerov-Cooley method or variation method. The Numerov-Cooley method is a general shooting type method capable potentially dealing with 1D potentials of any complexity. The variational methods are used in conjunction with different basis sets (FBR) or even with DVR. in the following, the general basis set input is introduced with different basis options explained in detail.
Basis set block
The structure of the Basis set block in the TROVE input is best illustrated using a specific example. Here we use the XY:sub:’2’ input as a typical example, such as SiH2:
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear', 'morse', range 0, 12, r 8, weight 2.0, points 1000,borders -0.8,1.40
1,'numerov','linear', 'morse', range 0, 12, r 8, weight 2.0, points 1000,borders -0.8,1.40
2,'numerov','linear', 'linear', range 0, 24, r 8, weight 1.0, points 1000,borders 10.0,160.0 deg
END
1. Sub-groups
Basis functions are combined into sub-groups by their symmetry equivalence and labeled with using an integer label, with the vibrational modes numbers with 1,2,3,… and the rotational basis is referenced to as 0.
The first line (mode) is for the rotational basis set functions and used specify the value of angular momentum 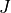 and also in some cases the coverage of the rotational quantum number
and also in some cases the coverage of the rotational quantum number 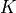 . In this entry,
. In this entry, JKtau is the type of the rotational basis set indicating the Wang symmetrisation of the type 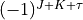 , see [23Yurchenko], which is currently the only option in TROVE. For
, see [23Yurchenko], which is currently the only option in TROVE. For 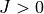 calculations the value of
calculations the value of Jrot is changed to of interest.
Other cards of the mode 0 include KMAX (Lmax) defining the maximal value of the rotational QN 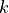 . It is used for the quasi-linear triatomics where the rotational QN is constrained to the vibrational angular momentum quantum number
. It is used for the quasi-linear triatomics where the rotational QN is constrained to the vibrational angular momentum quantum number 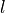 as
as 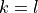 . Therefore, this card effectively defines the range of the vibrational angular momentum for quasi-linear triatomics,
. Therefore, this card effectively defines the range of the vibrational angular momentum for quasi-linear triatomics, 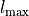 .
.
SiH2 has 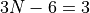 internal degrees of freedom and thus 3 vibrational basis functions are required. For this example, the first group
internal degrees of freedom and thus 3 vibrational basis functions are required. For this example, the first group 1 combines the two stretches SiH` 1 and SiH2 based on their symmetry equivalence: these two degrees of freedom transform through each other when acted upon with the C3v (M) symmetry operations. The sub-group ‘2’ is for the bending mode H-Si-H, which is a stand-alone degree of freedom that transforms independently from the sub-group 1. The grouping is used for producing symmetric combinations of basis functions and only coordinates symmetrically related should be grouped together. Details of this procedure are discussed in Theory and in [17YuYaOv].
2. Basis set method/type
For a given vibrational basis function row, the options are as follows. The first card specifies what the one-dimensional basis functions are. In this example they are numerically generated using the Numerov-Cooley method (Numerov card).
Basis set options
Numerov: basis functions are generated numerically using the Numerov-Cooley method.
harmonic: Analytic matrix elements or numerical values of the harmonic oscillator wavefunctions are used.
morse: basis functions and the corresponding matrix elements are based on the analytic expressions for the Morse polynomials.
Fourier: this basis set is introduced for modes with periodicity in periodic potential functions.
laguerre-k: is used in the direct connection with exact KEOs of triatomics, see sections about Frames.
sinrho-laguerre-k: is another basis set constructed for specific exact KEOs of triatomics, see sections about Frames.
3. Coordinates used to expand KEO
This card specifies how the kinetic energy operator is expanded. For the general linearised KEO, the default option is linear which refers to the expansion in terms of the displacements 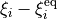 . For exact KEOs, which are nor general and different for different systems, frames, coordinates, this card defining the expansion types is usually associated with the KEO used.
. For exact KEOs, which are nor general and different for different systems, frames, coordinates, this card defining the expansion types is usually associated with the KEO used.
Here are the currently available options in TROVE:
‘Rational’: used for triatomic exact KEOs, which corresponds to the expansion of the stretching modes in terms of
(1st “order”),
(2nd “order”) and
(mixed term).
Automatic: the KEO constructed as part of the input (under construction).
Bond-Length: is similar to rational but independent for polyatomic molecules together with theAngletype. It has been used for the H2CS molecule [23MeOwTe].
Angletype is to expand the KEO in terms of the trigonometric combinations(1st order),
(2nd),
(3rd),
(4th),
and
, where
is a generic angle.
Dihedralis a part of thebond-length/angle/dihedraloption; it is used to introduce the following terms in the KEO:(1st),
(2nd),
(3rd),
(4th) and
(5th), where
is a generic dihedral mode.
4. Coordinates used to expand PEF
The next card gives the expansion coordinates for the potential. The available options are
linear, used for the displacement
morse, used to represent the expansion in the form of the Morse coordinateand a good option for the stretching coordinates.
cos(x)is to expand PEF in terms of, but can lead to singularities at around
and
.
5. Quantum number range
The numbers after range specify the range of vibrational quantum numbers of the one-dimensional functions to be used. For the example here, 0-12 is used for stretches and 0-24 for bends. The range should be consistent with the definition of the maximum polyad number used:
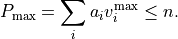
where 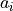 are the polyd coefficients (weights), defined in the next card.
are the polyd coefficients (weights), defined in the next card.
The borders card can be combined with the units cards, deg, Degree, Degrees, Bohr, for non-default units, e.g.
2,'laguerre-k','linear','linear', range 0,24, weight 1.0, points 2000, borders 0.,120.0 deg
6. Polyad weights
The number after weight (aka resc) gives the weighting of the vibrational quantum number for that coordinate in 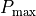 . Since the Si-H stretches here have a waiting of 2, it only makes sense to generate them from 0-12 if the polyad number is set to 24. The legacy aliases for
. Since the Si-H stretches here have a waiting of 2, it only makes sense to generate them from 0-12 if the polyad number is set to 24. The legacy aliases for weight are resc (resonance coefficients).
7-8. Integration points and borders
points and borders specify the number of points and the starting points for the Numerov-Cooley integration as the primary usage. Generating these one-dimensional functions is fast and so many points should be taken. The borders should be set far enough into the classically forbidden region of the potential such that the results are not sensitive to slightly larger or lower values. The units for borders are the same as those used that the potential was expanded in (Morse for stretches and angles in radians for bends in this example). For the Numerov-Cooley method, TROVE will check the numerical wavefunctions for their orthogonality and normalisation. If the latter properties are broken, TROVE will stop and suggest to increase the integration borders.
The second use of the coordinate grids defined by these tow cards is in the symmetrisation sampling procedure. Therefore these cards must be defined even for non Numerov-Cooley integration method.
The details of the primitive basis sets are given in the TROVE output file and will be discussed in output.
Other non-standard options
Reduced(aliasr): this card allows to reduce the expansion order of PEF when used to generate the basis set. It is sometimes more efficient for symmetry purposes to use a quadratic-type expansion in place of the full expansion with the order defined byPotOrder.
Periodicindicates that the potential is periodic and defines the periodicity. This property can be used to integrate the 1D problem on a smaller range and then extend by applying the periodic boundary conditions. Example:5,'fourier','linear', 'linear', range 0,17, weight 1.0, points 500, borders,0.d0,720.d0 deg, periodic 2
Lvib(Vib_Momentum) is used for systems where the basis set is constructed by diagonalising the vibrational angular momentum. The advantage of this construction scheme is that the basis set functions are assigned the vibrational angular momentum value
, where
As another example, it an be used to for spherical tops such as ammonia or phosphine to assign the vibrational basis and eignefunctions with he vibrational index
lviboption could help recover it.
Postprocess(post): this option is used to postprocess the contracted vibrational basis set generated on a reduced potential or Hamiltonian for the full PEF. It helps improve the basis set by re-optimising it. For example, for thelvib-constructed contracted basis functions, i.e. generated as eigenfunctions of
BASIS
0,'JKtau', jrot 0
1,'numerov','linear', 'morse', range 0, 4, weight 2.0, points 2000, borders -0.3,0.6
2,'numerov','linear', 'morse', range 0, 3, weight 1.0, points 1000, borders -0.5,0.75
2,'numerov','linear', 'morse', range 0, 3, weight 1.0, points 1000, borders -0.5,0.75
3,'harmonic','linear', 'linear',range 0, 6,r 2, weight 1.0, points 2000, borders -1.8,1.8 lvib post
3,'harmonic','linear', 'linear',range 0, 6,r 2, weight 1.0, points 2000, borders -1.8,1.8 lvib post
3,'harmonic','linear', 'linear',range 0, 6,r 2, weight 1.0, points 2000, borders -1.8,1.8 lvib post
3,'harmonic','linear', 'linear',range 0, 6,r 2, weight 1.0, points 2000, borders -1.8,1.8 lvib post
END
Here, the harmonic basis set was used for the sub-group 4 combing four linearised bending degrees of freedom of C2H2 as the basis for eigen-solving for the vibrational angular momentum (lvib). After the new wavefunctions are obtained as classified by , they are re-optimised (post) for the given by solving an eigenvalue problem for a reduced 4D Hamiltonian with a quadratic PEF (r 2).
Nocheckis used to suppress checking of the symmetry equivalence of the modes within the same sub-group. This is necessary for the modes which are dynamically symmetry equivalent. For example, when treating molecule CH3OH can be treat a C:subs:`3v`(M) molecule, the individual stretching CH modes are not equivalent at any fixed torsional configuration and would not be allowed in TROVE to be used for generating the basis sets. Instead, TROVE would choose the 1st mode at some reference torsional angle to generate a reference basis set and will used it for all three modes. For example:3, 'numerov', 'linear', 'morse', range 0, 4 , weight 1.0,points 1000,borders -0.4, 2.23 nocheck 3, 'numerov', 'linear', 'morse', range 0, 4 , weight 1.0,points 1000,borders -0.4, 2.23 nocheck 3, 'numerov', 'linear', 'morse', range 0, 4 , weight 1.0,points 1000,borders -0.4, 2.23 nocheck
Examples of Basis
H2O
BASIS
0,'JKtau', Jrot 0, krot 4
1,'numerov','rational', 'morse', range 0,12, r 8, weight 2.0, points 1000, borders -0.36,1.4
1,'numerov','rational', 'morse', range 0,12, r 8, weight 2.0, points 1000, borders -0.36,1.4
2,'laguerre-k','linear','linear', range 0,24, weight 1.0, points 2000, borders 0.,120.0 deg
END
NH3
BASIS
0,'JKtau', Jrot 2
1,'numerov','linear', 'morse', range 0, 4, r 8, weight 4.0, points 2000, borders -0.4,2.0
1,'numerov','linear', 'morse', range 0, 4, r 8, weight 4.0, points 2000, borders -0.4,2.0
1,'numerov','linear', 'morse', range 0, 4, r 8, weight 4.0, points 2000, borders -0.4,2.0
2,'harmonic','linear', 'linear', range 0,12, r 2, weight 2.0, points 9000, borders -1.90,1.91
2,'harmonic','linear', 'linear', range 0,12, r 2, weight 2.0, points 9000, borders -1.90,1.92
3,'numerov','linear', 'linear', range 0,12, r 8, weight 1.0, points 1000, borders -55.0, 55.0 deg
END
CH4
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear', 'morse', r 8, range 0, 0, weight 2.0, points 1000, borders -0.45,0.9
2,'numerov','linear', 'morse', r 8, range 0, 0, weight 2.0, points 1000, borders -0.45,0.9
2,'numerov','linear', 'morse', r 8, range 0, 0, weight 2.0, points 1000, borders -0.45,0.9
2,'numerov','linear', 'morse', r 8, range 0, 0, weight 2.0, points 1000, borders -0.45,0.9
3,'numerov','linear', 'linear',r 8, range 0, 0, weight 1.0, points 1000, borders -2.10,2.10 post
3,'numerov','linear', 'linear',r 8, range 0, 0, weight 1.0, points 1000, borders -2.10,2.10 post
3,'numerov','linear', 'linear',r 8, range 0, 0, weight 1.0, points 1000, borders -2.10,2.10 post
4,'harmonic','linear', 'linear',r 2, range 0, 6, weight 1.0, points 4000, borders -2.20,2.20 post
4,'harmonic','linear', 'linear',r 2, range 0, 6, weight 1.0, points 4000, borders -2.20,2.20 post
END
Rotational basis set
For the rotational motion, the standard rigid rotor wavefunctions are used, which are then symmetrised via the Wang-combinations (all expect for some special cases of the Td symmetry) in the 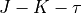 form as follows
form as follows
![\begin{split}
&|J,0,\tau\rangle = |J,0\rangle, \quad \tau = J\; {\rm mod}\; 2 , \\
&|J,K,\tau=0\rangle = \frac{1}{\sqrt{2}} \left[ |J,K,m\rangle + (-1)^{J+K} |J,-K,m\rangle \right],\\
&|J,K,\tau=1\rangle = \frac{i (-1)^{\sigma} }{\sqrt{2}} \left[ |J,K,m\rangle - (-1)^{J+K} |J,-K,m\rangle \right].
\end{split}](_images/math/f3b4224b970878e7471bf68c80106ccfaa0d8267.png)
Here 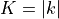 ,
, 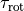 is the value associated with the parity of
and
is the value associated with the parity of
and 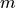 is omitted on the left-hand side for simplicity’s sake. is the rotational quantum number (
is omitted on the left-hand side for simplicity’s sake. is the rotational quantum number (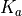 or
or 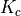 , depending on the orientation of the
, depending on the orientation of the  axis). The sign of is, however, not a physically meaningful quantity of a rotational eigen-state, but the parity is.
axis). The sign of is, however, not a physically meaningful quantity of a rotational eigen-state, but the parity is. 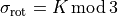 . The symmetry classification must be implemented for each molecule/symmetry case as part of the subroutines ML_rotsymmetry_<MOLECULE> for each nodule mol_<MOLECULE>.f90. For example, such an implementation for the rotational basis functions
. The symmetry classification must be implemented for each molecule/symmetry case as part of the subroutines ML_rotsymmetry_<MOLECULE> for each nodule mol_<MOLECULE>.f90. For example, such an implementation for the rotational basis functions 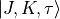 of C3v(M) are given by
of C3v(M) are given by
|
|
|
|
0 |
|
|
1 |
|
|
0 |
|
|
1 |
|
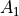
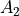
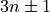
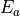
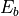
Once implemented, there is nothing else to be specified in the input file as far as the rotational basis set is concerned, except to set the value of :
BASIS
0,'JKtau', Jrot 15
.....
For a Td symmetry molecule like methane or silane, the symmetry-adapted rotational functions are obtained as linear combinations of
with 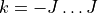 , spanning multiple values of . Therefore, the rotational quantum number can no longer be used for classification of these symmetrised rigid-rotor combinations. Instead they can be labelled as
, where
, spanning multiple values of . Therefore, the rotational quantum number can no longer be used for classification of these symmetrised rigid-rotor combinations. Instead they can be labelled as
, where 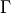 is the symmetry and
is the symmetry and 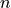 is a counting index:
is a counting index:
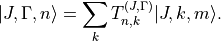
The coefficients 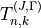 are generated based on the symmetry properties of the transformations of the Euler angles in
are generated based on the symmetry properties of the transformations of the Euler angles in 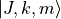 . All expansion coefficients required for this symmetry adaptation are generated automatically as part a numerical procedure. For this type of molecules, it is necessary to switch on by placing the card
. All expansion coefficients required for this symmetry adaptation are generated automatically as part a numerical procedure. For this type of molecules, it is necessary to switch on by placing the card rotsym euler inside the CONTRACTION block, e.g.
CONTRACTION
Npolyads 14
rotsym euler
model j=0
END