Frames and vibrational coordinates
Here we introduce different ingredients available for triatomic molecules, including
Molecular frames
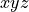 ;
;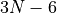 (
(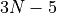 ) vibrational coordinates
) vibrational coordinates 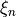 ;
;
For the linearised coordinates, the default frame is Eckart. The equilibrium structures, required for the definition of the linearised KEO and PEF, are chosen as the principal axis system (PAS). For the curvilinear KEOs, the frames are defined by the construction of the KEOs in their analytic representations.
Triatomics
XY2 type molecules
A molecule type is defined by the keyword MolType. For the XY2 example it is
MolType XY2
in the curvilinear KEO, it is common in TROVE to use the bisector frame for the XY2 molecules, with the 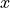 axis bisecting the bond angle and the
axis bisecting the bond angle and the  in the plane of the molecule, but other embeddings are possible. The PAS frame coincides with the bisector frame at the equilibrium or non-rigid reference configuration (i.e. symmetric). In TROVE, the definition of the frame is combined with the definition of the internal coordinates via the keywords
in the plane of the molecule, but other embeddings are possible. The PAS frame coincides with the bisector frame at the equilibrium or non-rigid reference configuration (i.e. symmetric). In TROVE, the definition of the frame is combined with the definition of the internal coordinates via the keywords transform or frame. In the following, these are described.
For quasi-linear triatomic molecules, it is also possible to use exact curvilinear KEO in implemented analytically, see below.
R-RHO-Z
R-RHO-Zis used for (quasi-)linear molecules of the XY2 type. it defined the curvilinear vibrational coordinates as the two bond angles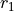 and
and 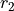 with the bending mode described by the angle
with the bending mode described by the angle 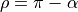 , where
, where 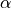 is the interbond angle (
is the interbond angle (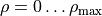 ). The exact orientation of the frame relative to the instantaneous position of the nuclei is critical for representing the dipole moment vectors.
). The exact orientation of the frame relative to the instantaneous position of the nuclei is critical for representing the dipole moment vectors.
For the rigid reference frame (REFER-CONF RIGID), the actual internal coordinates are the displacements of , and 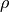 from the corresponding equilibrium:
from the corresponding equilibrium:
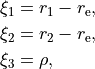
where 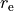 is the equilibrium bond length. If the non-rigid reference frame is used (
is the equilibrium bond length. If the non-rigid reference frame is used (REFER-CONF NON-RIGID), the bending mode is given on an equidistant grid, typically of 1000-2000 points, while the stretching modes are the displacements from the given point along the non-rigid reference frame, the latter is usually defined as the principal axes system with the bond length fixed to the equilibrium:
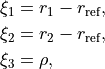
TROVE uses Z-matrix coordinates to build any user-defined coordinates. In this case, the Z=matrix is given by
ZMAT
S 0 0 0 0 31.97207070
H 1 0 0 0 1.00782505
H 1 2 0 0 1.00782505
end
Alternatively, the reference value of the bond length  can also vary with as e.g. in the minimum energy path (MEP) definition with being the optimised value at the given value of corresponding to the local energy minimum. In this case, the non-rigid frame must be defined using the
can also vary with as e.g. in the minimum energy path (MEP) definition with being the optimised value at the given value of corresponding to the local energy minimum. In this case, the non-rigid frame must be defined using the MEP block (see the corresponding section).
For the linearised coordinates type (COORDS Linear), the actual internal coordinates are the linearised versions of 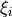 above. More specifically, for the
above. More specifically, for the non-rigid reference configuration, the bending coordinate :math`rho` is kept curvilinear on a grid of :math`rho_k` points as before, while the stretching coordinates are defined by linearly expanding :math`r_1` and :math`r_2` in terms of the Cartesian displacement around the corresponding reference values . In the Rigid case, the bending coordinate is also linearised.
The advantage of the linearised coordinates is that the corresponding KEO can be constructed on the fly as part of the TROVE generalised procedure as a Taylor type expansion. The main disadvantage however is that the approximate linearised KEO operator is less accurate than the (exact) curvilinear EKO. Besides, the convergence of the variational solution is also poorer for the linearised case (see [15YaYu]).
R-RHO-Z-ECKART
This Transform (frame) type is very similar to R-RHO-Z, but with the molecular frame define using the Eckart conditions.
R-ALPHA-Z
R-ALPHA-Zis very similar toR-RHO-Zwith the difference in the bending coordinate, which in the interbond angle in this case. In the Rigidreference configuration, it is a displacement from the equilibrium value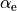 :
:
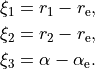
In the Non-rigid reference configuration, is given on a grid of points ranging from  to
to 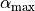 and including the equilibrium value. In the linearised
and including the equilibrium value. In the linearised Rigid case, the bending coordinated is defined as a linear expansion of at 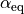 in terms of the Cartesian displacements.
in terms of the Cartesian displacements.
TROVE input example:
COORDS local (curvilinear coordinates)
frame r-rho-z (r1, r2, rho with the x parallel to the bisector)
MOLTYPE XY2
REFER-CONF non-RIGID (Reference configuration)
Note
The text in brackets is used for comments.
R-RHO-Z-M2-M3
A ‘bisecting’ XY2 frame used for isotopologies with slightly different masses of Y1 and Y2, for example O16CO17. Although this is an XYZ molecule, in this case it is formally treated as XY2 but with non-symmetric masses and the Cs symmetry, e.g.:
frame R-RHO-Z-M2-M3
MOLTYPE XY2
MOLECULE CO2
REFER-CONF non-RIGID
SYMGROUP Cs(M)
ZMAT
C 0 0 0 0 11.996709
O 1 0 0 0 16.995245
O 1 2 0 0 15.9905256
end
XYZ type molecules
The main embedding here is the ‘bond’-embedding, with the axis placed parallel to the bond Y-Z with a heavier atom Z comparing to X (second bond).
For molecules XYZ with comparable masses X and Z (e.g. in similar isotopologues), the bisector frames and associated frame can be used.
R1-Z-R2-RHO
This is a ‘bond’-embedding with the same vibrational coordinates as in R-RHO-Z and along the axis and in the negative direction of .

Frame R1-Z-R2-RHO: An XYZ type molecule and the embedding along and  with negative .
with negative .
The coordinates are given as above:
Here is an example of the Z-matrix for NNO.
ZMAT
N 0 0 0 0 14.00307401
N 1 0 0 0 14.00307401
O 1 2 0 0 15.994915
end
R1-Z-R2-ALPHA
This is another ‘bond’-embedding with the same vibrational coordinates as in R-ALPHA-Z.
R2-Z-R1-RHO
This is a ‘bond’-embedding with the along the axis and in the positive direction of , which is illustrated in the figure.
Exact KEO frames for triatomic molecules
There several exact, curvilinear KEO forms are available in TROVE for quasi-linear triatomic molecules, XY2 and XYZ. These KEOs are implemented in TROVE analytically, together with the corresponding matrix elements with the singularity resolution. These forms require a kinetic block in input with a reference to the specific frame. This is the difference with the linearised KEOs which use a general TROVE approach applicable for arbitrary molecules, except the linear ones. Exact KEO frames require that the COORDS card is set to LOCAL (aka CURVILINEAR), which stands for the curvilinear coordinates.
The associated kinetic expansion order KinOrder must be set to 2 in the following exact KEO. Here the expansion plays a formal role as this KEO i represented as a formal expansion of the 2nd order in terms of two stretches around the non-rigid reference configuration along the coordinate (see the rational expansion type in the basis set in the stretching subgroup 1).
Each KEO presented case is constructed to be used with the specific basis set configuration and usually also for a specific frame. These must be always used together.
KINETIC_XY2_EKE_BISECT
This is a bisector frame for curvilinear coordinates of an XY2 molecules with kinetic input block is given by
KINETIC
kinetic_type KINETIC_XY2_EKE_BISECT
END
It can be only used with the coordinates/frame type R-RHO-Z (see above), i.e. for the valence coordinates with as the bending angle (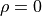 at the linear geometry), the basis set
at the linear geometry), the basis set laguerre-k and with the NON-RIGID reference configuration.
The laguerre-k basis functions are constructed using the Associated Laguerre polynomial with the factor 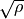 or
or 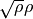 , depending if
, depending if 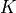 (rotational quantum number) is zero or not, respectively. The associated kinetic expansion order
(rotational quantum number) is zero or not, respectively. The associated kinetic expansion order KinOrder must be set to 2.
Here is an input example for this case for the C3 molecule:
KinOrder 2
COORDS local
frame r-rho-z
MOLTYPE XY2
REFER-CONF NON-RIGID
SYMGROUP C2v(M)
ZMAT
C 0 0 0 0 11.996709
C 1 0 0 0 11.996709
C 1 2 0 0 11.996709
end
BASIS
0,'JKtau', Jrot 0, krot 12
1,'numerov','rational', 'morse', range 0,30,r 8, weight 1.0, points 3000, borders -0.40,1.40
1,'numerov','rational', 'morse', range 0,30,r 8, weight 1.0, points 3000, borders -0.40,1.40
2,'laguerre-k','linear','linear', range 0,56, weight 1.0, points 10000, borders 0.,110.0 deg
END
KINETIC
kinetic_type KINETIC_XY2_EKE_BISECT
END
KINETIC_XYZ_EKE_BOND_SINRHO
This is a bond frame KEO constructed to work with the basis set type sinrho-laguerre-k. The associated frame is R1-Z-R2-RHO, for example for 12C12C13C, the corresponding TROVE input is as follows:
COORDS local (curvilinear)
TRANSFORM R-RHO-Z-M2-M3-BISECT (FRAME)
MOLTYPE XY2
REFER-CONF NON-RIGID
ZMAT
C 0 0 0 0 12.000000
C 1 0 0 0 13.003355
C 1 2 0 0 12.000000
end
KINETIC
kinetic_type KINETIC_XYZ_EKE_bisect
END
The associated symmetry is either Cs(M) or Cns(M):
symmetry Cs(M)
KINETIC_XY2_EKE_BISECT_SINRHO
This a similar to the basis set KINETIC_XY2_EKE_BISECT, which is introduced to work with the basis set sinrho-laguerre-k and only with this basis set. This basis set is constructed from the Associated Laguerre polynomial with the factor 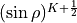 .
.
The associated TROVE configuration is as in the following input:
KinOrder 2
COORDS local
frame r-rho-z
MOLTYPE XY2
REFER-CONF NON-RIGID
BASIS
0,'JKtau', Jrot 0, krot 12
1,'numerov','rational', 'morse', range 0,30,r 8, resc 1.0, points 3000, borders -0.40,1.40
1,'numerov','rational', 'morse', range 0,30,r 8, resc 1.0, points 3000, borders -0.40,1.40
2,'sinrho-laguerre-k','linear','linear', range 0,56, resc 1.0, points 10000, borders 0.,110.0 deg
END
KINETIC
kinetic_type KINETIC_XY2_EKE_BISECT_SINRHO
END
KINETIC_XYZ_EKE_BISECT
For asymmetric triatomic molecules of type XYZ, there are several ways to orient the in-plane axes and at a general instantaneous geometry. The KINETIC_XYZ_EKE_BISECT KEO is constructed for the frame with the axis along the molecular bisector. The bisector XYZ frame is for asymmetric molecules XYZ with similar masses of Y and Z, i.e. when a bisector is a more natural description of the axis than a bond-frame. This KEO must be used with the correct XYZ-type bisector frames: R-RHO-Z-M2-M3-BISECT is used for general asymmetric molecules with similar masses of Y (M2) and Z (M3).
In principle, the KEO should fully define the configuration of the problem to solve and the associated frame type should not matter for the solution of the Schroedniger equation. The point where the choice of the frame becomes critical is when the dipoles are involved, which need to be re-defined into the correct frame. The actual transformation of the dipole is performed in the subroutine MLloc2pqr_xyz.
For example for 12C12C13C, the corresponding TROVE input is as follows:
COORDS local (curvilinear)
TRANSFORM R-RHO-Z-M2-M3-BISECT (FRAME)
MOLTYPE XY2
REFER-CONF NON-RIGID
ZMAT
C 0 0 0 0 12.000000
C 1 0 0 0 13.003355
C 1 2 0 0 12.000000
end
KINETIC
kinetic_type KINETIC_XYZ_EKE_bisect
END
The associated symmetry is either Cs(M) or Cns(M):
symmetry Cs(M)
KINETIC_XYZ_EKE_BOND
Is one of the bond-frames constructed for the XYZ type molecules (X is in the centre), with the axis along the instantaneous orientation of the bond (X-Y). Bond-frames are better suited for molecules with a light nucleus and the axis is assumed for the heavier nucleus. The associated frame is R1-Z-R2-RHO and the basis set type is laguerre-k:
KinOrder 2
COORDS local
frame r-rho-z
MOLTYPE XY2
REFER-CONF NON-RIGID
BASIS
0,'JKtau', Jrot 0, krot 12
1,'numerov','rational', 'morse', range 0,30,r 8, resc 1.0, points 3000, borders -0.40,1.40
1,'numerov','rational', 'morse', range 0,30,r 8, resc 1.0, points 3000, borders -0.40,1.40
2,'laguerre-k','linear','linear', range 0,56, resc 1.0, points 10000, borders 0.,110.0 deg
END
KINETIC
kinetic_type KINETIC_XYZ_EKE_BOND
END
KINETIC_XYZ_EKE_BOND-R2
This is the case of the bond-frame with along the bong (Z nucleus). The associated frames and basis sets are R2-Z-R1-RHO and laguerre-k, respectively. See the figure illustrating the R2-Z-R1-RHO frame above.
KINETIC_XYZ_EKE_BOND_SINRHO
This is a bond frame KEO constructed to work with the basis set type sinrho-laguerre-k. The associated frame is R1-Z-R2-RHO, for example for HCN, the corresponding TROVE input is as follows:
COORDS local (curvilinear)
TRANSFORM R1-Z-R2-RHO (FRAME)
MOLTYPE XY2
REFER-CONF NON-RIGID
ZMAT
C 0 0 0 0 12.00000000
N 1 0 0 0 14.00307401
H 1 2 0 0 1.00782503224
end
KINETIC
kinetic_type KINETIC_XYZ_EKE_BOND_SINRHO
END
The associated symmetry is either Cs(M) or Cns(M):
symmetry Cs(M)
Tetratomics
XY3 rigid molecules (PH3 type)
Linearized KEOs use the Eckart frame with the PAS at the equilibrium configuration. The latter has the axis along the axis of symmetry 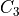 with the axis chosen in plane containing the X-Y1 bond and passing through .
with the axis chosen in plane containing the X-Y1 bond and passing through .
R-ALPHA
For the rigid XY3, like PH3, the logical coordinate choice of the valence coordinates consists of three bond lengths , , , 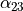 ,
, 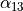 and
and 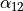 . For the linearised KEO, these valence are used to form the linearised coordinates in the same way as before (1st order expansion in terms of the Cartesian displacement). For the curvilinear KEO (
. For the linearised KEO, these valence are used to form the linearised coordinates in the same way as before (1st order expansion in terms of the Cartesian displacement). For the curvilinear KEO (local), the vibrational coordinates are then defined as displacement from the corresponding equilibrium (or non-rigid reference) values:
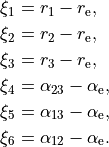
The underlying Z-matrix coordinates are defined using the following Z-matrix:
ZMAT
N 0 0 0 0 14.00307401
H 1 0 0 0 1.00782505
H 1 2 0 0 1.00782505
H 1 2 3 1 1.00782505
end
This representation has been used for PH3 [15SoAlTe], SbH3 [10YuCaYa], AsH3 [19CoYuKo], PF3 [19MaChYa].
XY3 non-rigid with umbrella motion (NH3 type)
MolType XY3
Consider the Ammonia molecule NH33 with a relatively small barrier to the planarity. The three bending angles are not suitable in this case as they cannot distinguish the two opposite inversion configurations above and below the planarity. Instead, an umbrella mode has to be introduced as one of the bending modes. An example of an umbrella coordinate is an angle between the symmetry axis and the bond X-Y, see Figure. It is natural to use the non-rigid reference configuration along the umbrella, inversion motion and build the KEO as an expansion around it. For two other bending modes, in principle one can use two inter-bond angles, e.g. 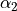 and
and 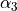 , two dihedral angles
, two dihedral angles 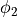 and
and 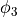 . However, for symmetry reasons, TROVE employs the symmetry-adapted bending pair
. However, for symmetry reasons, TROVE employs the symmetry-adapted bending pair 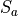 and
and 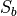 , defined as follows:
, defined as follows:
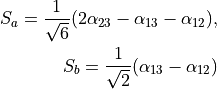
or
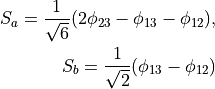
The umbrella mode for any instantaneous configuration of the nuclei is defined in TROVE as the angle between a trisector
Linearized KEOs use the Eckart frame with the PAS at the equilibrium configuration. The latter has the axis along the axis of symmetry with the axis chosen in plane containing the X-Y1 bond and passing through .
R-S-DELTA
For this frame case, the following valence-based coordinates are used:
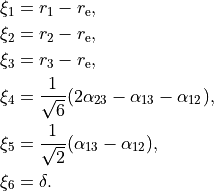
The umbrella mode :math:\delta is defined as an angle between the trisector and any of the bonds X-Y. The other 5 coordinates are then used to construct the corresponding linearised vibrational coordinates (see above) for the linearised (linear) representation.
ZXY2 (Formaldehyde type)
MolType ZXY2
The common valence coordinate choice for ZXY2 includes three bond lengths , two bond angles and a dihedral angle  . The latter can be treated as the reference for a non-rigid reference configuration in TROVE on a grid of
. The latter can be treated as the reference for a non-rigid reference configuration in TROVE on a grid of 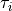 ranging from :math`[-tau_{0}ldots tau_{0}]`, while other 5 modes are treated as displacement from their equilibrium values at each grid point . The reference configuration is always in the principle axis sysetm, i.e. for each value of the book angle , TROVE solve the PAS conditions to reorient the molecule.
ranging from :math`[-tau_{0}ldots tau_{0}]`, while other 5 modes are treated as displacement from their equilibrium values at each grid point . The reference configuration is always in the principle axis sysetm, i.e. for each value of the book angle , TROVE solve the PAS conditions to reorient the molecule.
Apart from the standard linearised KEO, a curvilinear exact KEO has been recently introduced into TROVE. This is exactly the R-THETA-TAU type, detailed below.
R-THETA-TAU
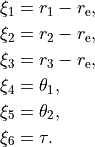
Rigid isotopologues of XY3 as ZXY2 type
The Z type can be used to define single or double deuterated isotopologues of an XY3 molecule such as a rigid PH3. For PDH2, we use R-THETA-TAU in combination with the Z-matrix given as follows:
ZMAT
P 0 0 0 0 14.00307401
D 1 0 0 0 2.01410178
H 1 2 0 0 1.007825032
H 1 2 3 2 1.007825032
end
Here, the equilibrium frame coinsides with the principle axis system with the axis in the plane contemning PD and bisecting the angle between two PH bonds.
For a PH2D type isotopologue, the Z-matrix is given by
ZMAT
P 0 0 0 0 14.00307401
H 1 0 0 0 1.007825032
D 1 2 0 0 2.01410178
D 1 2 3 2 2.01410178
end
Non-rigid isotopologues of XY3 as ZXY2 type
For non-rigid NH2D and NHD2, the choice of the non-rigid frame becomes important. During to the large amplitude motion, the frame can lead to a flip of the moments of inertia. In TROVE, the frame is chosen as the principle axis system (PAS), which, for most of the system, is straightforward to define. Therefore, frame and the internal coordinates are usually selected via a single keyword Transform. For non-rigid systems, however, due to accidental degeneracies of the moments of inertia, the PAS along the non-rigid path must be carefully constructed to prevent such flips. Besides, for the ammonia isotopologues, despite the structure ZXY2 being formally similar the structure of formaldehyde, the coordinates should be chosen as ammonia-like, not formaldehyde-like. Therefore, in this case we distinguish frame and transform. For NHD2, the following setting is used:
TRANSFORM R-S-DELTA
frame R1-Z-R2-X-Y
MOLTYPE XY3
REFER-CONF NON-RIGID
The valence coordinates is defined using the Zmat card as follows:
ZMAT
N 0 0 0 0 14.00307401
D 1 0 0 0 2.01410178
H 1 2 0 0 1.007825032
H 1 2 3 1 1.007825032
end
The coordinates type R-S-DELTA (card TRANSFORM) defines the internal coordinates the same as in the case of XY3, while the frame R1-Z-R2-X-Y places the axis containing the vector 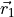 (ND), axis in the direction of the (NH1) and the symmetry plane to be
(ND), axis in the direction of the (NH1) and the symmetry plane to be 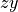 , see the figure. This non-rigid frame of NH2D is illustrated in the side figure, where the evolution of the PAS is shown. At the planar configuration, the
, see the figure. This non-rigid frame of NH2D is illustrated in the side figure, where the evolution of the PAS is shown. At the planar configuration, the 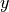 axis is normal to the plane with as the symmetry axis.
axis is normal to the plane with as the symmetry axis.
The card MOLTYPE XY3 means that all the associated transformation rules are implemented in the module mol_xy3.f90.
The same non-rigid frame is used for NHD2, now with the axis containing the vector (NH), axis in the direction of the (ND1) and the symmetry plane to be , see the figure. This non-rigid frame of NHD2 and the evolution of PAS is illustrated in the side figure with the Z-matrix given by
ZMAT
N 0 0 0 0 14.00307401
D 1 0 0 0 2.01410178
H 1 2 0 0 1.007825032
H 1 2 3 1 1.007825032
end
Although the definition of the frames is similar, the transformations of the corresponding PASs are very distinct.
A chain ABCD type molecule (hydrogen peroxide type)
MolType ABCD
R-ALPHA-TAU
The six internal coordinates for the frame R-ALPHA-TAU type consist of three stretching, two bending and one dihedral coordinates as given by
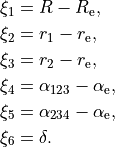
The non-rigid reference frame such that the axis bisects the dihedral angle.
For this embedding, in order to be able to separate the vibrational and rotational bases into a product form, it is important to the an extended range for the dihedral angle 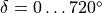 . Otherwise the eigenfunction is obtained double valued due to the axis appearing in the opposite direction to the two bonds after one
. Otherwise the eigenfunction is obtained double valued due to the axis appearing in the opposite direction to the two bonds after one 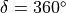 revolution.
revolution.
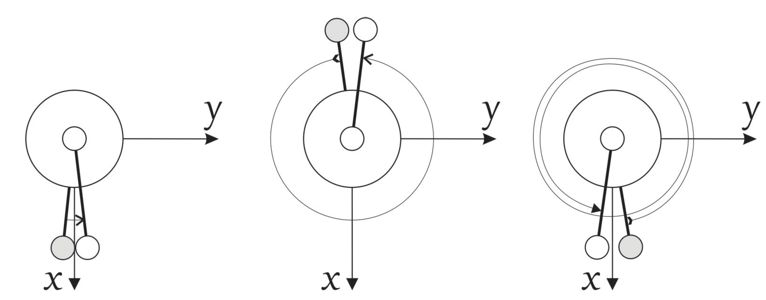
Principal axis system with an extended torsional angle for HOOH.
A minimum energy path (MEP) as a non-rigid reference configuration
In MEP, the 5 internal coordinate displacements are defined around 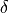 -dependent reference values. The latter are obtained as oprmised geometries by minimised molecule’s energy:
-dependent reference values. The latter are obtained as oprmised geometries by minimised molecule’s energy:
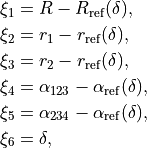
where :math: the MEP values are given by a parameterised expansion, for example
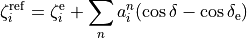
where 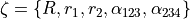 .
.
Five-atomic molecules
The XY4 molecule (Td) and the XY4 type
MolType XY4
The frame for the tetrahedral molecule XY4 spanning the Td(M) symmetry group is chosen with the axes orthogonal to the faces of the box containing the molecule with the four atoms  at its vertices, as shown in the figure,
with the Cartesian coordinates at equilibrium given by
at its vertices, as shown in the figure,
with the Cartesian coordinates at equilibrium given by
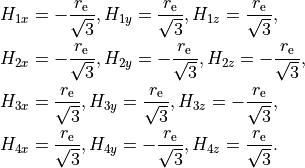
TRANSFORM R-ALPHA
The tetrahedral five-atomic molecule XY4 has 9 vibrational degrees of freedom. For a semi-rigid molecule (i.e. ignoring any isomerisation that can occur at higher energies), they can be characterised by four bond lengths  and six inter-bond angles
and six inter-bond angles 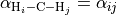 . For the equilibrium value of the tetrahedral angle ,
. For the equilibrium value of the tetrahedral angle , 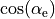 =
= 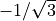 which explains the factor
which explains the factor 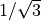 in the definition of the Cartesian coordinates.
There should, however, be only 9 independent vibrational degrees of freedom in a 5 atomic molecule. One of the inter-bond angles
in the definition of the Cartesian coordinates.
There should, however, be only 9 independent vibrational degrees of freedom in a 5 atomic molecule. One of the inter-bond angles 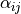 is redundant as there should be only five independent bending vibrations, with the following redundancy condition:
is redundant as there should be only five independent bending vibrations, with the following redundancy condition:
(1)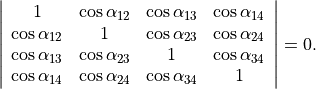
XY4 belongs to the Td(M) molecular symmetry group, which consists of five irreducible representations, 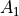 ,
, 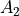 ,
, 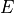 ,
, 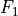 and
and 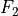 . One way to define independent bending modes is to reduce the six inter-bond angles to five symmetry-adapted irreducible combinations, which, together with four bond lengths
. One way to define independent bending modes is to reduce the six inter-bond angles to five symmetry-adapted irreducible combinations, which, together with four bond lengths 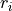 form nine independent vibrational modes as follows: four stretches
form nine independent vibrational modes as follows: four stretches
(2)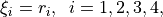
two -symmetry bends
(3)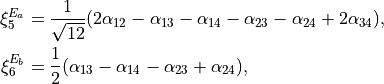
and three 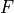 -symmetry bends
-symmetry bends
(4)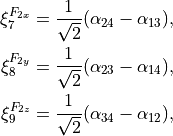
where the corresponding symmetries of the bending modes are indicated.
The stretching modes can also be in principle combined into symmetry-adapted coordinates in Td(M):
(5)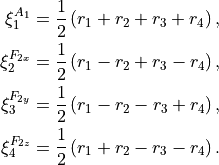
ZXY3 (Methyl Chloride type)
MolType ZXY3
For the ZXY3 type molecule we use valence coordinates consisting of four bond lengths 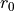 , (
, (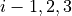 ), three bond angles
), three bond angles 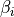 and two symmetry adapted dihedral coordinates constructed from three dihedral angles
and two symmetry adapted dihedral coordinates constructed from three dihedral angles 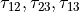 , where
, where 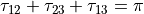 . This is a
. This is a rigid type, where all coordinates are treated as displacements from the corresponding equilibrium values. Currently, only the standard linearised KEO is available in TROVE.
TRANSFORM R-BETA-SYM
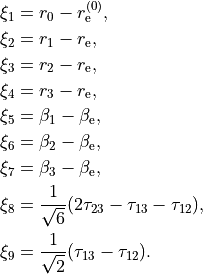
The Z-matrix coordinates (underlying basic TROVE coordinates) are as given by the Z-matrix rules:
ZMAT
C 0 0 0 0 12.000000000
Cl 1 0 0 0 34.968852721
H 1 2 0 0 1.007825035
H 1 2 3 0 1.007825035
H 1 2 3 4 1.007825035
end
are as follows:
- ,
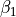
- ,
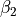 ,
, - ,
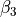 ,
,
where alpha_{12}` and are interbond angles between the bonds X-Yi. The Z-matrix coordinates are transformed
to :math:`tau_{12}, tau_{23}, tau_{13} ` via the following trigonometric rules:
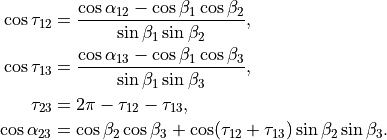
The CH3D molecule (C3v) of the ZXY3 type
TRANSFORM R-BETA-SYM
This is a similar to the Methyl Chloride molecule type (MOLTYPE ZXY3) in terms of the symmetry properties, although being a methane-deuterated product. The same coordinate Transform type TRANSFORM R-BETA-SYM as for fZXY3 can be used for CH3D with the following setting:
TRANSFROM R-BETA-SYM
MOLTYPE ZXY3
but with the PES of methane (see Potential energy functions).
Six-atomic molecules
The C2H4 molecule and the C2H4 type
MolType C2H4
C2H4_2BETA_1TAU
The internal coordinates are defined using the following 12 valence coordinates: 5 stretching (molecular bond) coordinates, 4 bending (inter-bond angles) and 3 dihedral coordinates, with the last mode as a book angle describing the relative motion of two moieties:
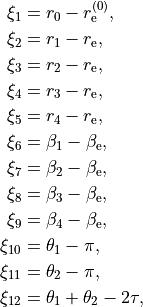
where
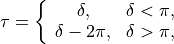
This type can be used both for rigid and non-rigid molecule types. The non-rigid coordinate is 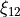 in the latter case.
in the latter case.