Benchmarks and examples
A set of TROVE benchmarks for a number of typical systems can be found here
PH3
This PH3 benchmark is to produce a small line list for phosphine (PH3) using a relatively large primitive basis set.
It includes the following input files:
ph3_P18_step1.inp
ph3_P18_step2.inp
ph3_P18_step3_J0.inp
ph3_P18_step3_J1.inp
ph3_P18_step3_J2.inp
ph3_P18_step3_J3.inp
ph3_P18_step3_J4.inp
ph3_P18_step3_J5.inp
ph3_P18_step3_J10.inp
ph3_P18_step3_J50.inp
ph3_P18_step4_intensity.inp
covering the following steps:
Step 1: Produce the initial smaller
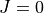 HAMILTONIAN matrix and diagonalize it using
HAMILTONIAN matrix and diagonalize it using DSYEV(step1)Step 2: Convert to
representation; prepare matrix elements for 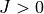 calculations (step2)
calculations (step2)- Step 3: Generate a
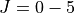 10, 50 energies and eigenfunctions (step3-step9) by diagonalising three double real, symmetric matrices for each J using DSYEV.
10, 50 energies and eigenfunctions (step3-step9) by diagonalising three double real, symmetric matrices for each J using DSYEV. Each calculation is independent, but depends on the checkpoints produced at steps 1-2. It requires checkpoints from steps 1-2.
- Step 3: Generate a
Step 4: Intensity calculations between J=0-5 states (ph3_P18_step4_intensity.inp). It will depend on the checkpoints produced at steps 1-3.
Memory and time requirements:
Step1: 84 Gb 27032.7
Step2: 9.9 Gb 1220.9 s
Step3: 0.8 Gb 2.6 s
Step4: 6.8 Gb 73 s
Step5: 7.8 Gb 198 s
Step6: 8.4 Gb 502 s
Step7: 9.0 Gb 830 s
Step8: 9.6 Gb 1449 s
Step9: 19.3 Gb 9032 s
Step10: 32.5 Gb 12096 s
Step_intensity: 89 Gb 1592 s
Disk space used: 300 Gb
Here 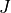 is the rotational angular momentum.
is the rotational angular momentum.
There are three matrices to diagonalise for each symmetry 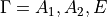 . The dimensions of the matrices are given approximately by
. The dimensions of the matrices are given approximately by
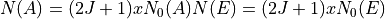
where 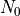 are the dimensions of the
are the dimensions of the 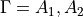 and
and 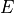 matrices at ,
matrices at , 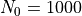 and 2000, respectively (approximately).
and 2000, respectively (approximately).
The first step in this case is relatively expensive, 84 Gb and 27032.7 s on a 16 processor workstation.
The input file has the structure described in detail in the following.
Stand-alone keywords, memory, verbosity level, expansion order of KEO and PEF, sparse methods.
mem 64 gb
verbose 5
KinOrder 6 (Max order in the kinetic energy expansion)
PotOrder 8 (Max order in the potential energy expansion)
Natoms 4 (Number of atoms)
Nmodes 6 (Number of modes = 3*Natoms-6)
sparse
More stand-alone keywords:
Finite difference steps, general coordinates type (
linear-ised orlocal), TROVE coordinates, Molecule type, the molecule name (only for printing), reference configuration:dstep 0.01 COORDS linear TRANSFORM R-ALPHA MOLTYPE XY3 MOLECULE PH3 REFER-CONF RIGID
Molecular symmetry group
SYMGROUP C3v(M)
Primitive basis block:
PRIMITIVES
Npolyads 18 (Number of primitive polyads)
enercut 80000. (energy cut for the primitive basis set)
END
Contracted basis block:
CONTRACTION
Npolyads 18 (Number of contracted polyads)
enercut 80000.0 (energy cut for the contracted basis set)
degeneracy 1e-3 (threshold used to define degenerate energies)
coeff_thresh 1e-14 (threshold used to define symmetry)
enercut_matelem 10000.0
exp_coeff_thresh 1.0d-6
END
Eigensolver (diagonalizer) block:
DIAGONALIZER
syev
enermax 15000
end
Zmatrix block defining the structure and masses:
ZMAT
P 0 0 0 0 30.9737620
H 1 0 0 0 1.00782505
H 1 2 0 0 1.00782505
H 1 2 3 0 1.00782505
end
Control block:
control step 1 external end
Basis set block defining the order of the primitive basis set functions and how their grouped into the contracted basis sets:
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear', 'morse', range 0, 9, resc 2.0, points 2000, borders -0.5,2.10
1,'numerov','linear', 'morse', range 0, 9, resc 2.0, points 2000, borders -0.5,2.10
1,'numerov','linear', 'morse', range 0, 9, resc 2.0, points 2000, borders -0.5,2.10
2,'numerov','linear', 'linear', range 0,18, resc 1.0, points 2000, borders -1.6,1.30
2,'numerov','linear', 'linear', range 0,18, resc 1.0, points 2000, borders -1.6,1.30
2,'numerov','linear', 'linear', range 0,18, resc 1.0, points 2000, borders -1.6,1.30
END
Equilibrium structure block:
EQUILIBRIUM
re 1 1.41182210
re 1 1.41182210
re 1 1.41182210
alphae 0 93.3685 deg
alphae 0 93.3685 deg
alphae 0 93.3685 deg
end
“Special” parameters block, mostly used to define the Morse stretching parameters and in some cases harmonic frequencies or Laguerre structural parameter.
SPECPARAM
beta 0 1.8000000
beta 0 1.8000000
beta 0 1.8000000
END
Potential energy function block:
POTEN
NPARAM 109 (obsolete)
POT_TYPE poten_xy3_morbid_10
COEFF list (powers or list)
VE 0 0.00000000000000E+00
FA1 1 -0.34350288027976E+03
FA2 1 0.28963515335650E+06
FA3 1 -0.66897678674464E+06
FA4 1 0.26097579162907E+07
......
end
Dipole moment function block
DIPOLE
rank 3
NPARAM 127 0 0
DMS_TYPE XY3_MB
COEFF list (powers or list)
COORDS linear
Order 2 2 2
parameters
charge 0 0.00000000
order 0 4.00000000
alphae 0 93.40000000
re14 0 1.41200000
beta 0 1.00000000
gamma 0 0.00000000
delta 0 0.00000000
mu0 0 0.33369443
F1 0 -1.05840018
F3 0 0.13117597
F4 0 0.17351923
F5 0 -0.21359267
.....
end
H3+
This is another pyramid-type molecule but with a non-rigid umbrella degree of freedom treated using a non-rigid reference configuration and linearised coordinates to represent the KEO. The PEF is empirical from [20YuTeMi]. The calculation steps are as in the example of PH3.
SiH2 Refinement
Two directories SiH2-1 and SiH2-2 provide examples of the TROVE refinement for a rigid XY2 type system:
./trove.x <SiH2_step1.inp > SiH2_step1.out
./trove.x <SiH2_step2.inp > SiH2_step2.out
./trove.x <SiH2_step3_J0.inp > SiH2_step3_J0.out
./trove.x <SiH2_step3_J1.inp > SiH2_step3_J1.out
./trove.x <SiH2_step3_J2.inp > SiH2_step3_J2.out
./trove.x <SiH2_step4.inp > SiH2_step4.out
./trove.x <SiH2_step5.inp > SiH2_step5.out
H2CO
H2CO-1: A typical TROVE line list pipeline for H2CO treated as a rigid molecule using a non-exact KEO (linearised coordinates).
In order to run this benchmark on COSMOS submit using, e.g.
The actual running script is run_trove_benchmark.csh for the following jobs:
./trove.x <file1.inp >file1.out
./trove.x <file2.inp >file2.out
./trove.x <file3.inp >file3.out
./trove.x <file4.inp >file4.out
./trove.x <file5.inp >file5.out
./trove.x <file6.inp >file6.out
./trove.x <file7.inp >file7.out
./trove.x <file8.inp >file8.out
./trove.x <file_intensity.inp > file_intensity.out
with the steps as in the PH3 example.
HCN
An example of a linear molecule with an exact KEO.
The TROVE package must be compiled with pot_HCN.f90 (module pot_user), which defines the potential energy function of HCN as the general type and a PEF from [16MaKyPo].
This benchmark contains the following jobs:
./trove.x <HCN_step1.inp > HCN_step1.out
./trove.x <HCN_step2.inp > HCN_step2.out
./trove.x <HCN_step3_J0.inp > HCN_step3_J0.out
./trove.x <HCN_step3_J1.inp > HCN_step3_J1.out
./trove.x <HCN_step3_J2.inp > HCN_step3_J2.out
This TROVE calculation represents the following steps:
Step 1: Produce the initial smaller J=0 HAMILTONIAN matrix and diagonalize it using DSYEV (
HCN_step1.inp)Step 2: Convert to
HCN_step2.inp)Step 3: Generate a
energies and eigenfunctions (
HCN_step3_J<0,1,2>.inp) by diagonalising double real, symmetric matrices for each