Symmetry
TROVE uses the Molecule Symmetry Group [98BuJe] to classify the ro-vibrational states, motion, coordinates etc. The symmetries are defined in the molecule.f90 file.
In order to specify the symmetry, the keyword SYMGROUP should be given anywhere in the input file outside any sections, but before the DIAGONALIZER section, e.g.
SYMGROUP D2H(M)
Here the molecular symmetry group is D2h(M).
C(M)
C(M) is the simplest symmetry which means no symmetry with one irreducible representation (irrep) 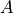 .
.
Cs(M)
Cs(M) is the second simplest symmetry group with two irreducible representations 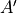 and
and 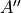 :
:
SYMGROUP Cs(M)
It is usually used for non-symmetric planar molecules such as triatomics XYZ.
C2v(M)
C2v(M) is a molecular symmetry group consisting of 4 irreps: 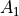 ,
, 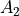 ,
, 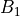 ,
, 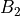 . The meaning of these irreps depends on the molecule as well as its embedding. The characters are shown in the following table
. The meaning of these irreps depends on the molecule as well as its embedding. The characters are shown in the following table
|
(12) |
|
(12)* |
|
|---|---|---|---|---|
|
1 |
1 |
1 |
1 |
|
1 |
1 |
-1 |
-1 |
|
1 |
-1 |
-1 |
1 |
|
1 |
-1 |
1 |
-1 |
where 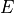 , (12),
, (12), 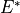 and (12)* are the 4 point group operations. Typical C2v(M) molecules are XY2 (e.g. water, H2S), ZXY2 (e.g. formaldehyde.)
and (12)* are the 4 point group operations. Typical C2v(M) molecules are XY2 (e.g. water, H2S), ZXY2 (e.g. formaldehyde.)
Symmetry properties of the vibrational coordinates
TROVE uses the symmetry properties of the vibrational coordinates, i.e. how they transform upon applying the symmetry operations, to build symmetry adapted vibrational basis functions. The symmetrisation method is described in [17YuYaOv]. In the following, we show how the coordinates, we show these transformation properties for the corresponding coordination frames implemented in TROVE.
R-RHO-Z, R-ALPHA-Z
Coordinates |
|
(12) |
|
(12)* |
|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
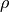
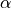
C3v(M)
C3v(M) is a molecular symmetry group consisting of 3 irreps: , , and 6 operations:
|
(123) |
(23)* |
|
|---|---|---|---|
(132) |
(12)* |
||
(23)* |
|||
|
1 |
1 |
1 |
|
1 |
1 |
-1 |
|
2 |
-1 |
0 |
It can be used for the molecules PH3 [15SoAlTe], SbH3 [10YuCaYa], AsH3 [19CoYuKo], PF3 [19MaChYa], CH3Cl [18OwYaTe], CH3F, isotopologue CDH3 etc.
Coordinate transformation properties
R-ALPHA
This is a rigid frame with 6 valence coordinates 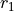 ,
, 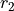 ,
, 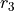 ,
, 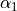 ,
, 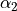 and
and 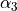 .
.
Coordinates |
|
(123) |
(132) |
(23)* |
(13)* |
(12)* |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D3h(M)
D3h(M) is a molecular symmetry consisting of 6 irreps: 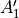 ,
, 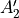 ,
, 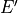 ,
, 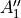 ,
, 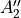 ,
, 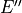 and 12 operations:
and 12 operations:
|
(123) |
(23) |
|
(123)* |
(23)* |
|
|---|---|---|---|---|---|---|
(132) |
(12) |
(132)* |
(12)* |
|||
(23) |
(23)* |
|||||
|
1 |
1 |
1 |
1 |
1 |
1 |
|
1 |
1 |
-1 |
1 |
1 |
-1 |
|
2 |
-1 |
0 |
2 |
-1 |
0 |
|
1 |
1 |
1 |
-1 |
-1 |
-1 |
|
1 |
1 |
-1 |
-1 |
-1 |
1 |
|
2 |
-1 |
0 |
-2 |
1 |
0 |
The D3h(M) group has been used for NH3 [10CoYuTe], CH3 [19AdJeYa].
This is a non-rigid frame with 3 valence stretching coordinates , , , a symmetry adapted bending vector 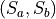 and an umbrella coordinate
and an umbrella coordinate 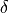 , where
, where
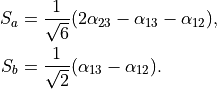
The transformation properties of the stretching coordinates are given by
Coordinates |
|
(123), (123)* |
(321), (321)* |
(23),(23)* |
(12),(12)* |
(13),(13)* |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The bending vector transforms as follows
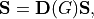
where 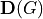 are 2x2 transformation matrices given by
are 2x2 transformation matrices given by
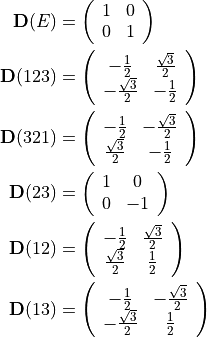
The operations with inversion have the same matrices, 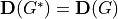 .
.
Finally, the umbrella coordinate transform as follows
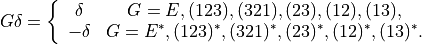
Td(M)
Td(M) is a molecular symmetry group is used for the methane-like molecules, CH4 [14YuJe], SiH3 [17OwYuYa]. It consists of 5 irreps and 24 symmetry operations spanning 5 classes:
|
(123) |
(14)(23) |
(1423)* |
(23)* |
|
|---|---|---|---|---|---|
Elements |
1 |
8 |
3 |
6 |
6 |
|
1 |
1 |
1 |
1 |
1 |
|
1 |
1 |
1 |
-1 |
-1 |
|
2 |
-1 |
2 |
0 |
0 |
|
3 |
0 |
-1 |
1 |
-1 |
|
3 |
0 |
-1 |
-1 |
1 |
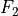
Numerical symmetry group Dnh
As a numerical application of linear-molecule symmetry properties, described by the D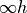 point group, lower-order symmetry groups Dnh(M) with finite
point group, lower-order symmetry groups Dnh(M) with finite 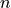 can be used. Character tables and irreducible representation transformation matrices are implemented in TROVE for Dnh(M) groups with arbitrary - values. These groups can subsequently be used in the construction of symmetry-adapted ro-vibrational basis functions for solving the Schrödinger equations of linear molecules. HCCC is an example of a linear molecule of the D point group symmetry for which Dnh(M) can be used in symmetrisations.
can be used. Character tables and irreducible representation transformation matrices are implemented in TROVE for Dnh(M) groups with arbitrary - values. These groups can subsequently be used in the construction of symmetry-adapted ro-vibrational basis functions for solving the Schrödinger equations of linear molecules. HCCC is an example of a linear molecule of the D point group symmetry for which Dnh(M) can be used in symmetrisations.
Table: Irreducible representations for the Dnh(M) groups and their characters under the generating operations 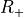 ,
, 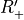 and
and 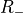 ( is even).
( is even).
Dnh |
|
|
|
|
|---|---|---|---|---|
( |
|
|
|
|
|
1 |
1 |
1 |
1 |
|
1 |
1 |
1 |
-1 |
|
1 |
-1 |
1 |
1 |
|
1 |
-1 |
1 |
-1 |
|
2 |
|
2 |
0 |
|
1 |
1 |
-1 |
1 |
|
1 |
1 |
-1 |
-1 |
|
1 |
-1 |
-1 |
1 |
|
1 |
-1 |
-1 |
-1 |
|
2 |
|
-2 |
0 |
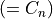
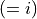
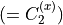
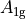
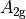
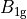
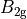
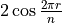
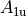
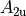
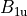
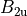
where for 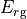 and
and 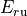 ,
,  = 1, 2, dots,
= 1, 2, dots, 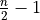 .
.
Table: Irreducible representations for the Dnh(M) groups and their characters under the generating operations , and ( is odd).
Dnh |
|
|
|
|
|---|---|---|---|---|
( |
|
|
|
|
|
1 |
1 |
1 |
1 |
|
1 |
1 |
1 |
-1 |
|
2 |
|
2 |
0 |
|
1 |
1 |
-1 |
1 |
|
1 |
1 |
-1 |
-1 |
|
2 |
|
-2 |
0 |
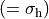
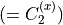
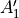
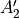
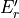
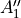
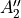
where for 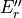 , = 1, 2, dots,
, = 1, 2, dots, 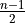 .
.
The following table gives the correspondence between the 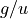 (gerade/ungerade) notation of the irreps of Dnh (even ) and the
(gerade/ungerade) notation of the irreps of Dnh (even ) and the 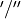 notation of the irreps of Dnh (odd ), based on
notation of the irreps of Dnh (odd ), based on 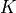 (the absolute value of the projection, in units of
(the absolute value of the projection, in units of 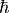 , onto the molecule-fixed
, onto the molecule-fixed  -axis of the rotational angular momentum).
-axis of the rotational angular momentum).
Artificial symmetries (AEM)
The concept of artificial molecular symmetries was introduced in [21MeYuJe].
C2vn(AEM)
Linear molecules usually represent a special case in rotational-vibrational calculations due to a singularity of the kinetic energy operator that arises from the rotation about the  (the principal axis of least moment of inertia, becoming the molecular axis at the linear equilibrium geometry) being undefined. Assuming the standard ro-vibrational basis functions, in the
(the principal axis of least moment of inertia, becoming the molecular axis at the linear equilibrium geometry) being undefined. Assuming the standard ro-vibrational basis functions, in the 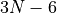 approach, of the form
approach, of the form 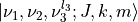 , tackling the unique difficulties of linear molecules involves constraining the vibrational and rotational functions with
, tackling the unique difficulties of linear molecules involves constraining the vibrational and rotational functions with 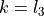 , which are the projections, in units of , of the corresponding angular momenta onto the molecular axis. These basis functions are assigned to irreps of the C
, which are the projections, in units of , of the corresponding angular momenta onto the molecular axis. These basis functions are assigned to irreps of the C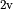 (M) molecular symmetry group. This, in turn, necessitates purpose-built codes that specifically deal with linear molecules. In the present work, we describe an alternative scheme and introduce an (artificial) group that ensures that the condition
(M) molecular symmetry group. This, in turn, necessitates purpose-built codes that specifically deal with linear molecules. In the present work, we describe an alternative scheme and introduce an (artificial) group that ensures that the condition 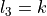 is automatically applied solely through symmetry group algebra. The advantage of such an approach is that the application of symmetry group algebra in ro-vibrational calculations is ubiquitous, and so this method can be used to enable ro-vibrational calculations of linear molecules in polyatomic codes with fairly minimal modifications.
is automatically applied solely through symmetry group algebra. The advantage of such an approach is that the application of symmetry group algebra in ro-vibrational calculations is ubiquitous, and so this method can be used to enable ro-vibrational calculations of linear molecules in polyatomic codes with fairly minimal modifications.
In TROVE an alternative scheme is implemented as an (artificial) group that ensures that the condition is automatically applied solely through symmetry group algebra. The advantage of such an approach is that the application of symmetry group algebra in ro-vibrational calculations is ubiquitous, and so this method can be used to enable ro-vibrational calculations of linear molecules in polyatomic codes with fairly minimal modifications. To this end, we construct an artificial molecular symmetry group C2vn(AEM), which consists of one-dimensional (non-degenerate) irreducible representations and use it to classify vibrational and rotational basis functions according to 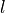 and
and 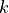 . This extension to non-rigorous, artificial symmetry groups is based on cyclic groups of prime-order. Opposite to the usual scenario, where the form of symmetry adapted basis sets is dictated by the symmetry group the molecule belongs to, here the symmetry group C2vn(AEM) is built to satisfy properties for the convenience of the basis set construction and matrix elements calculations. We believe that the idea of purpose-built artificial symmetry groups can be useful in other~applications.
. This extension to non-rigorous, artificial symmetry groups is based on cyclic groups of prime-order. Opposite to the usual scenario, where the form of symmetry adapted basis sets is dictated by the symmetry group the molecule belongs to, here the symmetry group C2vn(AEM) is built to satisfy properties for the convenience of the basis set construction and matrix elements calculations. We believe that the idea of purpose-built artificial symmetry groups can be useful in other~applications.
Examples of character tables for 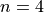 are given in Table below
are given in Table below
C2vn(AEM) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
|
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
|
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
|
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
|
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
|
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
|
1 |
1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
-1 |
1 |
|
1 |
-1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
|
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
|
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
|
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
|
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
1 |
1 |
-1 |
|
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
1 |
1 |
|
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
1 |
-1 |
|
1 |
1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
1 |
1 |
-1 |
-1 |
|
1 |
-1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
1 |
-1 |
-1 |
1 |
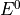
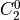
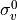
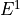
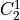
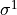
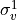
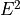
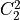
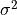
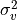
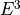
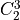
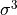
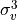
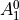
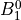
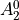
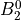
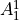
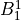
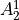
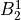
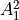
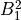
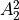
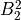
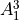
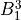
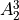
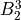
IN TROVE outputs, three-character labels 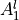 ,
, 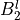 ,
, 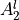 and
and 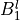 for
for 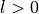 are replaced with the two-character shortcuts
are replaced with the two-character shortcuts  ,
, 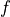 ,
, 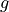 and
and 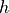 , respectively:
, respectively:
C2vn(AEM) |
TROVE output |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
… |
|
|
|
|
|
|
|
|
|
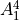
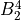
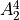
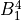
How to use C2vn(AEM)
An artificial symmetry C2vn(AEM) is invoked via the following card places anywhere (but before the diagonalizer section):
SYMGROUP C2vn 18
Here the integer number 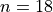 corresponds to the maximal value of the vibrational angular momentum
corresponds to the maximal value of the vibrational angular momentum 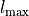 and therefore to the maximal value of the rotational quantum number due to the constraint
and therefore to the maximal value of the rotational quantum number due to the constraint 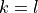 used for linear triatomic molecules (case 3N-6). This number muster coincide with the value of the
used for linear triatomic molecules (case 3N-6). This number muster coincide with the value of the k or kmax cards in the BASIS block (rotational basis line), e.g.
BASIS
0,'JKtau', Jrot 0, krot 18
1,'numerov','rational', 'morse', range 0,20, r 8, resc 3.0, points 2000, borders -0.3,0.90
2,'numerov','rational', 'morse', range 0,36, r 8, resc 1.5, points 3000, borders -0.4,0.90
3,'laguerre-k','linear','linear', range 0,48, r 8, resc 1.0, points 12000, borders 0.,120.0 deg
END
Cns(AEM)
The artificial symmetry group Cns(AEM) consists of one-dimensional, real irreps, which are contracted to correlate with irreps of C . The irreps of Cns(AEM) are labelled as
. The irreps of Cns(AEM) are labelled as 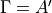 and irreps with an extra subscript (see Table below), e.g.,
and irreps with an extra subscript (see Table below), e.g., 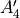 . For example, a vibrational function with
. For example, a vibrational function with 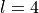 and transforming as in Cs would be assigned the symmetry in Cns(AEM). The 0-superscripted irreps are the only physical irreps, matched to and of Cstogether with the corresponding characters of each element, while all irreps of are non-physical, i.e. “artificial”. The full description of this case is given in [24YuMeTe].
and transforming as in Cs would be assigned the symmetry in Cns(AEM). The 0-superscripted irreps are the only physical irreps, matched to and of Cstogether with the corresponding characters of each element, while all irreps of are non-physical, i.e. “artificial”. The full description of this case is given in [24YuMeTe].
C4s |
Cs |
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|
|
|
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
|
|
1 |
-1 |
1 |
-1 |
1 |
-1 |
1 |
-1 |
|
1 |
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
|
|
1 |
-1 |
-1 |
1 |
1 |
-1 |
-1 |
1 |
|
|
1 |
1 |
1 |
1 |
-1 |
-1 |
-1 |
-1 |
|
|
1 |
-1 |
1 |
-1 |
-1 |
1 |
-1 |
1 |
|
|
1 |
1 |
-1 |
-1 |
-1 |
-1 |
1 |
1 |
|
|
1 |
-1 |
-1 |
1 |
-1 |
1 |
1 |
-1 |
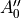
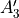
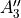
In TROVE outputs, 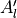 and
and 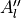 are replaced with
are replaced with 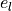 and
and 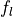 for compactness.
for compactness.
The effects of the Cns(AEM) group operations on the coordinates is as follows: all operations leave the vibrational coordinates invariant; the 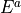 operations (in the notation of Table above leave the rotational functions invariant while the
operations (in the notation of Table above leave the rotational functions invariant while the 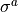 operation has the same effect as the
operation has the same effect as the 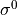 operation.
operation.
How to use Cns(AEM)
An artificial symmetry Cns(AEM) is invoked via the following card places anywhere (but before the diagonalizer section):
SYMGROUP Csn 18
The integer number corresponds to the maximal value of the vibrational angular momentum and therefore to the maximal value of the rotational quantum number due to the constraint used for linear triatomic molecules (case 3N-6). This number muster coincide with the value of the k or kmax cards in the BASIS block (rotational basis line) (see above).