Molecules
In order to define a Hamiltonian for an arbitrary molecular systems the following ingredients are required:
Molecular frame
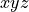 defining the rotation of the molecule via the positions of the three Euler angles
defining the rotation of the molecule via the positions of the three Euler angles 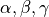 ;
; (
( ) vibrational coordinates
) vibrational coordinates 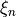 , internal degrees of freedom defining the molecular vibrations;
, internal degrees of freedom defining the molecular vibrations;Kinetic energy operator (KEO) in a sum-of-products form. In TROVE it has to be an expansion in terms of some 1D functions of
, their conjugate momenta and rotational angular momenta 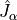 ;
;Potential energy function (PEF). In TROVE, it is also an expansion in terms of some 1D functions of
.For intensity calculations, 3D dipole moment functions, also as expansion in terms of some 1D functions of
.
In this chapter a list is given of all of the molecules which have been studied using TROVE. This serves two purposes: as a summary of the development of TROVE over the years and as a reference for adding further molecules. As discussed in Adding new molecules, setting up a new molecule in TROVE is fairly straightforward if a molecule of the same symmetry and structure has already been implemented.
The list of molecules are given in roughly chronological order with the relevant references. Details are given of the symmetry, basis sets used, coordinates used, the analytical forms of the PES and DMS and the frequency ranges of linelists if they were calculated.
Here we introduce different ingredients available for triatomic molecules, including
Molecular frames
;- () vibrational coordinates ;
Kinetic energy operators (KEO);
Potential energy functions (PEF);
For intensity calculations, 3D dipole moment functions.
Hydrogen sulfide, SiH2
Symmetry: 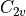
Coordinates: Linearized coordinates. 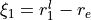 ,
, 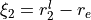 and
and 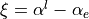 .
.
Coordinate to expand kinetic energy: 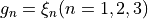 .
.
Coordinates to expand Potential energy: 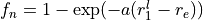
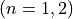 ,
, 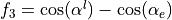
Primitive basis set: Numerov generated for all coordinates
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 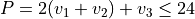
Potential energy function: POTEN_XY2_MORSE_COS with a refined potential represented in terms of Morse coordinates and 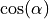 .
.
Dipole moment surface expansion: Ab initio DMS of the type xy2_pq_coeff.
Reference: [TROVE]
The TROVE input for step 1 is illustrated below.
mem 64 gb
KinOrder 6
PotOrder 8
Natoms 3
Nmodes 3
SYMGROUP C2v(M)
verbose 5
dstep 2.0e-03
COORDS linear
FRAME r-rho-z
MOLTYPE XY2
REFER-CONF NON-RIGID
PRIMITIVES
Npolyads 24
END
CONTRACTION
Npolyads 24
sample_points 60
sample_attempts 500
symm_toler 1e-5
END
DIAGONALIZER
SYEV
enermax 18000
end
ZMAT
Si 0 0 0 0 27.97692654
H 1 0 0 0 1.007825032
H 1 2 0 0 1.007825032
end
control
step 1
external
end
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear', 'morse', range 0, 12, r 8, resc 2.0, points 3000,borders -0.8,1.40
1,'numerov','linear', 'morse', range 0, 12, r 8, resc 2.0, points 3000,borders -0.8,1.40
2,'numerov','linear', 'linear', range 0, 24, r 8, resc 1.0, points 3000,borders 10.0,160.0 deg
END
EQUILIBRIUM
re 9 1.5144017558
re 9 1.5144017558
alphae 9 92.00507388 DEG
end
SPECPARAM
a-Morse 0 0.127050746200E+01
a-Morse 0 0.127050746200E+01
END
POTEN
NPARAM 102
compact
POT_TYPE POTEN_XY2_MORSE_COS
COEFF list (powers or list)
RE13 0.15144017558000E+01
ALPHAE 0.92005073880000E+02
AA 0.12705074620000E+01
B1 0.50000000000000E+06
B2 0.50000000000000E+05
G1 0.15000000000000E+02
G2 0.10000000000000E+02
V0 0.00000000000000E+00
F_0_0_1 -0.11243403302598E+02
F_1_0_0 -0.94842865087918E+01
F_0_0_2 0.17366522840412E+05
F_1_0_1 -0.25278354456474E+04
F_1_1_0 0.20295521820240E+03
F_2_0_0 0.38448640879698E+05
.....
....
end
DIPOLE
dimension 3
NPARAM 72 99 0
compact
TYPE xy2_pq_coeff
COEFF list (powers or list)
COORDS linear linear linear
Orders 6 6 6
dstep 0.005 0.005 0.005
Parameters
re 0.152000000000E+01
alphae 0.945000000000E+02
f_1_0_0 -0.170274198034E+01
f_1_0_1 -0.122791150585E+00
f_2_0_0 -0.519187500441E+00
f_1_0_2 0.185415937182E+00
f_2_0_1 0.715740118118E+00
f_2_1_0 -0.147662628115E+00
f_3_0_0 0.598556914831E+00
.....
re 0.152000000000E+01
alphae 0.945000000000E+03
a 0.000000000000E+00
dummy 0.000000000000E+00
xp(1) 0.176547582678E+01
x0x0x1 -0.492245503195E+01
x1x0x0 -0.193070832496E+01
x0x0x2 0.900424248416E+01
x0x2x0 0.114484321174E+01
x1x0x1 -0.116840841811E+01
x2x0x0 -0.101953882061E+01
x0x0x3 -0.152151621639E+02
.....
.....
end
A short description of the keywords, cards and sections used is as follows.
Mem: Maximal memory value available for the job ingb,mborkb. TROVE uses an internal book keeping of the memory usage and will stop if it is large than thememvalue.
KinOrder: Expansion order of the KEO.
PotOrder: Expansion order of the PEF.
Natoms: Number of atoms (nuclei).
Nmodes: Number of modes or degrees of freedom(here
).
SYMGROUP: Molecular symmetry group.
verbose: Verbosity level controlling amount of information in the standard output.
dstep: numerical difference step size used in finite differences (Angstrom or radian).
COORDS: type of the coordinate,linear(linearised) orlocal(curvilinear).
FRAME: Molecular frame.
MOLTYPE: The type of molecule (XYZ, XY2, XY3, XY4, ZXY3, etc).
REFER-CONF: reference configuration,RIGIDorNON-RIGID.
PRIMITIVES: block defining parameters of the primitive bases.
Npolyads: Maximal number of polyads.
CONTRACTION: Block defining parameters of the contracted basis set.
Npolyads: Maximal number of polyads in the contracted basis.
sample_points: number of sampling points in the symmetrisation procedure.
sample_attempts: number of symmetrisation attempts.
symm_toler: Numerical tolerance used in symmetrisation.
DIAGONALIZER: Block defining the diagonaliser (eigensolver) as well as its options (number of roots, maximal energy etc).
SYEV: LAPACK Eigensolver type DSYEV.
enermax: Maximal energy (cm-1).
ZMAT: Z-matrix block defining the Z-matrix coordinates and nuclear (atomic) masses.
control: Control block (see TROVE Quick start).
Basis: Basis set block (See Basis sets).
EQUILIBRIUM: Equilibrium values of the molecule geometries in terms of the Z-matrix coordinates.
SPECPARAM: Special parameters used to define the coordinate to expand PEF, e.g. the Morse parameter.
POTEN: Potential block (see Potential energy functions).
DIPOLE: Dipole moment block (orexternalfield block).
CO2
This is a linear triatomic molecule for which a number of exact KEOs implemented in TROVE.
Molecular type (MOLTYPE): XY2.
Symmetry: there are two types of symmetries available for symmetric quasi-linear triatomic molecules, molecular symmetry group (EM) and Artificial extended molecular symmetry 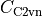 (AEM).
(AEM).
Coordinates: curvilinear (local).
Coordinates type (TRANSFORM): r-rho-z: 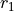 ,
, 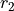 , and
, and 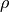 .
.
Frame:  axis is along the molecule at the linear configuration, with the
axis is along the molecule at the linear configuration, with the 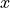 -axis oriented in-plane as a bisector at any instantaneous bend configuration of nuclei. The definition of the frame in this case is directly linked to the coordinate type
-axis oriented in-plane as a bisector at any instantaneous bend configuration of nuclei. The definition of the frame in this case is directly linked to the coordinate type r-rho-z is therefore omitted from the input.
It is based on seven internal coordinates defined using the following Z-matrix (using nuclear masses):
ZMAT
C 0 0 0 0 11.996709
O 1 0 0 0 15.9905256
O 1 2 0 0 15.9905256
end
Kinetic energy operator
KEO: exact, constructed using the bisector frame and formally expanded in terms of 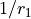 ,
, 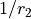 to the 2nd order about the non-rigid reference frame.
to the 2nd order about the non-rigid reference frame.
KEO type (kinetic_type): KINETIC_XY2_EKE_BISECT
Kinetic energy expansion order: 2
Primitive basis set: Numerov-generated for coordinates and and the Laguerre-k for :
BASIS
0,'JKtau', J 0, krot 40
1,'numerov','rational', 'morse', range 0,36, resc 1.0, points 1000, borders -0.40,1.0
1,'numerov','rational', 'morse', range 0,36, resc 1.0, points 1000, borders -0.40,1.0
2,'laguerre-k','linear','linear', range 0,64, resc 1.0, points 3000, borders 0.,160.0 deg
END
Here the card krot 40 indicates the highest vibrational angular momentum and the rotational quantum number 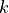 (See Chapter about Basis sets). rational stands for the expansion in terms of or . Modes 1 and 2 are combined into sub-group 1.
(See Chapter about Basis sets). rational stands for the expansion in terms of or . Modes 1 and 2 are combined into sub-group 1.
Basis set: Polyad scheme with 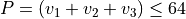 , :math:` v_1 leq 36`, :math:` v_2 leq 36`, :math:` v_3 leq 64`.
, :math:` v_1 leq 36`, :math:` v_2 leq 36`, :math:` v_3 leq 64`.
Spectroscopic Model UCL-4000
This model was used to produce the UCL-4000 line list for CO [23MeOwTe].
[23MeOwTe].
PEF type (POT_TYPE): poten_co2_ames1. This is the PEF Ames1 by Huang et al. [17HuScFr].
Potential expansion order: 8 using the PEF poten_xy3_morbid_10. Expansion order (PotOrder) is 12.
Coordinates to expand Potential energy: Morse (see basis ) for stretching coordinates while the angle is treated explicitly.
Potential energy function: Refinement of published potential [09YuBaYa].
Dipole moment surface expansion: TYPE DIPOLE_AMES1, AMES1 DMS by Huang et al. [17HuScFr].
For the spectroscopic model UCL-4000, a sample input file can be found at exomol.com, see UCL-4000 spectroscopic model.
Non-symmetric isotopologue 18CO16
The basic model is the same as for CO2 with some differences.
Molecular type (MOLTYPE): XY2 (same as before).
Symmetry: there are two types of symmetries available for non-symmetric quasi-linear triatomic molecules, molecular symmetry group  (EM) and Artificial extended molecular symmetry
(EM) and Artificial extended molecular symmetry  (AEM).
(AEM).
Coordinates: curvilinear (local).
Frame and associated coordinates type (TRANSFORM): R-RHO-Z-M2-M3. It uses the same valence coordinates as before, , , and . In the linear configuration, the frame is the axis along the molecule oriented from atom 1 to atom 2 (i.e. from mass 2 to mass 3, see Z-matrix), which is the same orientation as r-rho-z and also with the bisecting axis .
The following Z-matrix (using nuclear masses) is used:
ZMAT
C 0 0 0 0 11.996709
O 1 0 0 0 17.99477097
O 1 2 0 0 15.99052598
end
Kinetic energy operator
KEO: exact, constructed using the bisector frame and formally expanded in terms of , to the 2nd order about the non-rigid reference frame.
KEO type (kinetic_type): KINETIC_XYZ_EKE_BISECT. It is also a bisector-frame KEO but developed for a non-symmetric molecule XYZ
Kinetic energy expansion order: 2
Primitive basis set has the same structure as for the XY:
:
- BASIS
0,’JKtau’, Jrot 0, krot 16 1,’numerov’,’rational’, ‘morse’, range 0,36,r 8, resc 1.0, points 4000, borders -0.40,1.55 2,’numerov’,’rational’, ‘morse’, range 0,36,r 8, resc 1.0, points 4000, borders -0.40,1.55 3,’laguerre-k’,’linear’,’linear’, range 0,56, resc 1.0, points 12000, borders 0.,150.0 deg
END
with the main difference that all three modes are treated independently.
Spectroscopic Model UCL-4000
The spectroscopic model (PEF and DMF) are the same used to produce the UCL-4000 line list for CO [23MeOwTe].
KOH
This is a linear triatomic molecule for which a number of exact KEOs implemented in TROVE.
Molecular type (MOLTYPE): XY2.
Symmetry: (EM) and or .
Coordinates: curvilinear (local).
Coordinates type (TRANSFORM): R2-Z-R1-RHO: , , and .
Frame: axis is along atom 2 (vector ), with the -axis oriented in-plane at any bend configuration of nuclei and positive for atom 2 (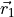 ). The definition of the frame in this case is directly linked to the coordinate type
). The definition of the frame in this case is directly linked to the coordinate type R2-Z-R1-RHO is therefore omitted from the input.
Z-matrix:
ZMAT
O 0 0 0 0 15.99491463
K 1 0 0 0 38.9637074
H 1 2 0 0 1.007825035
end
Kinetic energy operator
KEO: exact, formally expanded in terms of , to the 2nd order about the non-rigid reference frame.
KEO type (kinetic_type): KINETIC_XY2_EKE_BISECT_sinrho. It is linked to the basis set type sinrho-laguerre-k used for the bending mode.
BASIS
0,'JKtau', Jrot 0, krot 10
1,'numerov','rational', 'morse', range 0,20, resc 2.0, points 1000, borders -0.5 ,2.0
2,'numerov','rational', 'morse', range 0,20, resc 2.0, points 1000, borders -0.35,0.8
3,'sinrho-laguerre-k','linear','linear', range 0, 40, resc 1.0, points 3000, borders 0.,180.0 deg
END
Basis set: Polyad scheme with 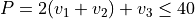 .
.
Spectroscopic Model OYT4
This model was used to produce the OYT4 line lists for KOH and NaOH [21OwTeYu].
PEF type (POT_TYPE): poten_xyz_tyuterev.
Potential expansion order: ab initio; expansion order = 8.
Dipole moment surface expansion: TYPE ` DIPOLE_PQR_XYZ`` with parameters from ab initio calculations.
A sample input file can be found at KOH_OYT4_model_TROVE.inp, which uses a different coordinate type from the model used for OYT4,
OCS
TBP
Ammonia, NH3
Ammonia is intrinsically a non-rigid system with a low barrier to the planarity and spectroscopically non-negligible tunneling splitting of about 0.78 cm -1. Therefore it must be treated using the non-rigid frame and an associated 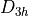 (M) group symmetry.
(M) group symmetry.
Molecular type (MOLTYPE): XY3.
Symmetry: (M)
Coordinates: linearised (Linear).
Coordinates type (TRANSFORM): r-s-delta. It is based on seven internal coordinates defined using the following Z-matrix:
ZMAT
N 0 0 0 0 14.00307401
H1 1 0 0 0 1.00782503223
H2 1 2 0 0 1.00782503223
H3 1 2 3 1 1.00782503223
end
As usual, it defines three stretching coordinates 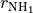 ,
, 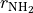 and
and 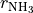 . For the angles, note that atom 4 has the “dihedral” type 1. For this type, TROVE introduced four angles (one of which is redundant): the first two angles are valence between atoms H2 and H1 (
. For the angles, note that atom 4 has the “dihedral” type 1. For this type, TROVE introduced four angles (one of which is redundant): the first two angles are valence between atoms H2 and H1 (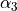 ), H3 and H1 (
), H3 and H1 (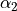 ). Angle 3 is also valence, between H3 and H2 (
). Angle 3 is also valence, between H3 and H2 (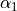 ). Angle 4 is dihedral between two planes:
). Angle 4 is dihedral between two planes: 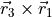 and
and 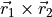 . These 4 angles are then used to construct two degenerate non-redundant bending coordinates:
. These 4 angles are then used to construct two degenerate non-redundant bending coordinates:
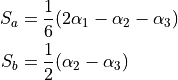
The last coordinates is an angle 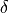 between the trisector and any of the bond vectors.
between the trisector and any of the bond vectors.
The valence coordinates are then used to define 5 linearised coordinates:
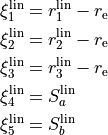
while the sixth coordinate is curvilinear 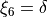 .
.
The Equilibrium block in the case of 7 coordinates is given by 7 equilibrium values:
EQUILIBRIUM
re 0 1.0116
re 0 1.0116
re 0 1.0116
alphae 0 106.719 deg
alphae 0 106.719 deg
alphae 0 106.719 deg
taue 0 0.385722379
end
This seven internal coordinates scheme provide a better numerical stability.
Kinetic energy operator
KEO: non-exact, constructed using the Sorensen procedure as an expansion about the non-rigid reference frame.
Frame: Non-rigid, Eckart conditions, follows the umbrella motion for a rigid stretches and equal angles.
Coordinate to expand kinetic energy: 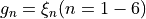
Kinetic energy expansion order: 6
Primitive basis set: Numerov generated for coordinates 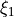 ,
, 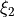 ,
, 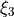 and
and 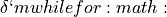 xi_4` and
xi_4` and 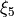 , the Harmonic oscillator basis gives more stable symmetry adaptation:
, the Harmonic oscillator basis gives more stable symmetry adaptation:
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear', 'morse', range 0, 8, r 8, resc 4.0, points 2000, borders -0.4,2.0
1,'numerov','linear', 'morse', range 0, 8, r 8, resc 4.0, points 2000, borders -0.4,2.0
1,'numerov','linear', 'morse', range 0, 8, r 8, resc 4.0, points 2000, borders -0.4,2.0
2,'harmonic','linear', 'linear', range 0,34, r 2, resc 2.0, points 9000, borders -1.90,1.91
2,'harmonic','linear', 'linear', range 0,34, r 2, resc 2.0, points 9000, borders -1.90,1.92
3,'numerov','linear', 'linear', range 0,34, r 8, resc 1.0, points 1000, borders -55.0, 55.0 deg
END
Spectroscopic Model BYTe
Potential expansion order: 8 using the PEF poten_xy3_morbid_10.
Coordinates to expand Potential energy: Morse for stretching coordinates, angles themselves for bends.
Basis set: Polyad scheme with 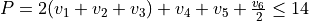 .
.
Potential energy function: Refinement of published potential [09YuBaYa].
Dipole moment surface expansion: DMF XY3_SYMMB. For the BYTe line list, an ab initio DMS was computed at the CCSD(T)/aug-cc-pVQZ level of theory [09YuBaYa].
Results: Hot line list called BYTe. BYTe is applicable for temperatures up to 1500 K. It comprises of 1138 323 351 transitions in the frequency range from 0 to 12 000 wavenumbers, constructed from 1373 897 energy levels below 18 000 wavenumbers having J values  36.
36.
Note
Apart from BYTe, ammonia was used to develop TROVE itself, specifically for the J=0 contraction and refinement methods. The BYTe line list remains important for astronomical applications but will also soon be joined by an even more accurate line list from the work of Coles et al. [10CoYuTe].
Reference: [09YuBaYa], [11YuBaTe], [10CoYuTe].
For BYTe, a sample input file can be found at exomol.com, see BYTe spectroscopic model.
Spectroscopic model CoYuTe
Potential energy function: general as defined in a stand-alone pot-user module pot_NH3_Roman.f90. PEF was expanded to the 8th order using the internal linearised coordinates.
Basis set: Polyad scheme with 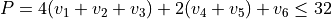 .
.
Dipole moment surface expansion: Same in BYTe.
A sample input file defining the spectroscopic model can be found at CoYuTe spectroscopic model.
Methyl cation, CH3+
Symmetry:
Coordinates: Linearized coordinates. 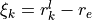
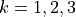 for vibrational coordinates, two symmetrized bending variables
for vibrational coordinates, two symmetrized bending variables 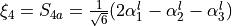 and
and 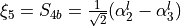 and an out of plane vibration coordinate
and an out of plane vibration coordinate 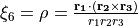 . See paper for details.
. See paper for details.
Coordinate to expand kinetic energy:
Coordinates to expand Potential energy: 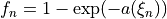
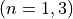 for stretching coordinates,
for stretching coordinates, 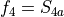 ,
, 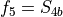 for two bending modes and
for two bending modes and 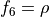 .
.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 6
Polyad scheme: 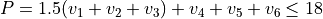
Potential energy function: Published potential.
Dipole moment surface expansion: N/A
Results: 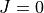 vibrational energy levels up to 6000 cm-1.
vibrational energy levels up to 6000 cm-1.
Note
This was also used as a test example in the original TROVE paper. The coordinate scheme employed is similar to that for Ammonia (see below).
Reference: [TROVE]
Phosphine, PH3 (rigid)
We consider phosphine as a rigid molecule with the tunneling splitting ignored.
Symmetry: 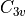
Coordinates type: R-ALPHA with the three stretching and three inter-bond bending coordinates.
Primitive basis set: Numerov generated for all coordinates.
Polyad scheme: 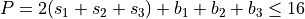 plus some additions, see paper.
plus some additions, see paper.
Z-matrix
The internal valence coordinates (required in construction of the linearised ones) are defined using the following Z-matrix:
ZMAT
P 0 0 0 0 30.9737620
H1 1 0 0 0 1.00782505
H2 1 2 0 0 1.00782505
H3 1 2 3 0 1.00782505
end
Here, atom 4 has the “dihedral” type 0, which is used for the interbond angles. In this case, the first two angles are between atoms H2 and H1 (), H3 and H1 () and the last angle is defined also as an interbond angle between H3 and H2 ().
Kinetic energy operator
Frame: rigid with Eckart
KEO: non-exact based on linearised coordinates
Kinetic energy expansion order: 6
Potential energy function
PEF type (POT_TYPE): poten_xy3_morbid_10.
Potential expansion order: 8.
Potential energy function: CCSD(T)/aug-cc-pV(Q+d)Z) ab initio energies fitted to polynomial expansion and refined to the HITRAN energies up to 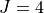 .
.
Dipole moment function
Dipole type (DMS_TYPE): XY3_MB
Dipole moment surface expansion: CCSD(T)/aug-cc-pVTZ ab initio dipole data fitted to polynomial expansion.
Result
Line list: SAlTY, complete for up to 1500 K. All states up to 18000 cm-1 included, up to 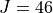
A sample input file can be found at exomol.com, see SAlTY spectroscopic model.
References: [13SoYuTe], [15SoAlTe].
Phosphine, PH3 (non-rigid)
For PH3, tunneling splitting via the umbrella motion may exist (as for NH3) may exist but has yet to be detected [16SoYuTe]. In order to treat phosphine as a non-rigid, the same setup as for NH3 can used with the symmetry group (M).
KEO: non-exact, constructed using the Sorensen procedure as an expansion about the non-rigid reference frame.
Molecular type (MOLTYPE): XY3.
Symmetry: (M)
Frame: Non-rigid, Eckart conditions, follows the umbrella motion for a rigid stretches and equal angles.
Coordinates type (TRANSFORM): r-s-delta.
Primitive basis set: Numerov generated for the stretched and the umbrella mode and the Harmonic basis for the two degenerate dihedral coordinates:
BASIS
0,'JKtau', Jrot 0
1,'numerov' ,'linear', 'morse', range 0, 7, r 8, resc 8.0, points 2000, borders -0.5,1.70
1,'numerov' ,'linear', 'morse', range 0, 7, r 8, resc 8.0, points 2000, borders -0.5,1.70
1,'numerov' ,'linear', 'morse', range 0, 7, r 8, resc 8.0, points 2000, borders -0.5,1.70
2,'harmonic','linear', 'linear', range 0,24, r 2, resc 4.0, points 12000, borders -3.00,3.01
2,'harmonic','linear', 'linear', range 0,24, r 2, resc 4.0, points 12000, borders -3.00,3.02
3,'numerov' ,'linear', 'linear', range 0,90, r 8, resc 0.6, points 10000, borders -80.0, 80.0 deg, period -2
END
Here, the numerical grid of the umbrella mode ranges from negative to positive angles with a planer structure in the middle. The card period -2 helps to build a symmetry adapted tunneling basis containing both the symmetric and asymmetric wavefunctions with a relatively large numerical grid of 5000 points.
As in the case of Ammonia, the transform type r-s-delta uses internally seven coordinates, , , 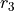 , , , , and the umbrella coordinate
, , , , and the umbrella coordinate 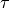 . Accordingly, the
. Accordingly, the Equilibrium requires seven values for the corresponding equilibrium values:
EQUILIBRIUM
re 1 1.41182210
re 1 1.41182210
re 1 1.41182210
alphae 0 93.3685 deg
alphae 0 93.3685 deg
alphae 0 93.3685 deg
taue 0 0.573251573522
end
Sulfur trioxide, SO3
The model is essentially the same as used for Ammonia (see above) and described in [13UnTeYu] and [16UnTeYu].
Symmetry: (M).
Kinetic energy expansion order: 6
Coordinates type: r-s-delta
PEF: A refined PES of type poten_xy3_morbid_10.
Potential expansion order: 8
Polyad scheme: 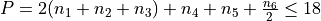 .
.
Potential energy function: CCSD(T)-F12b/aug-cc-pVTZ-F12 + scalar relativistic corrections and DBOCs ab initio energies fitted to polynomial expansion of symmetrised coordinates. Refined using 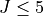 experimental energies.
experimental energies.
Dipole moment surface expansion: The same type as for Ammonia (XY3_SYMMB) based on ab initio calculations at the same levels as for PES. Fitted using the SMB representation.
Results: Linelist complete up to 5000 cm-1 for temperatures up to 800 K.
Note
As SO3 has a large moment of inertia, many 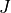 s need to be included. Up to
s need to be included. Up to 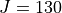 was included for a complete linelist at 800 K. For calculating this large, special procedures were used as discussed in the paper.
was included for a complete linelist at 800 K. For calculating this large, special procedures were used as discussed in the paper.
An example of the TROVE input file for the SO3 calculations using the UYT2 model can be found at UYT2 spectroscopic model.
References: [13UnTeYu], [16UnTeYu].
Methane, CH4
Spectroscopic Model 10to10
The model is described in [14YuJe].
KEO: non-exact, expanded in terms of linearised coordinates around a rigid reference geometry
REFER-CONF RIGID
Symmetry: 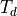
Frame: Eckart.
Coordinates: Type R-SYM, linearised coordinates obtained from the following valence coordinates:
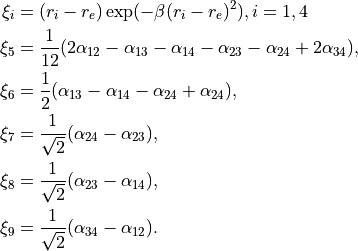
where 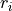 are the bond-lengths and
are the bond-lengths and 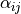 are the interbond angles, also complimented by redundancy conditions (see [14YuJe]).
are the interbond angles, also complimented by redundancy conditions (see [14YuJe]).
Coordinate to expand kinetic energy: 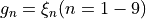 , linearised coordinates.
, linearised coordinates.
Coordinates to expand Potential energy: 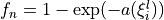
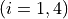 for stretching coordinates and
for stretching coordinates and 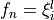
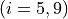 for bending coordinates.
for bending coordinates.
Primitive basis set: Numerov generated for stretching coordinates, harmonic oscillator basis for bends.
Kinetic energy expansion order: 6
PEF: type general implemented as a stand alone (pot_user) module pot_ch4.f90. Original PEF CCSD(T)-F12c/aug-cc-pVQZ-F12 + DK relativistic corrections ab initio was refined to experimental (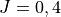 ) data from the HITRAN 2008 database.
) data from the HITRAN 2008 database.
Potential expansion order: 8
Polyad scheme: 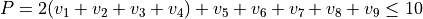 .
.
DMF: type general included in the same module pot_ch4.f90. Dipole moment surface expansion: CCSD(T)-F12c/aug-cc-pVTZ-F12 ab initio points were fit using polynomial of symmetrised coordinates which is then expressed in symmetrised molecular bond (SMB) representation, see [[13YuTeBa]]_.
Results: 10to10 linelist complete for up to 1500 K. All states up to 18000 cm-1 included, up to J = 39.
Note
This describes the 10to10 calculation which was based on a previous calculation for lower frequencies. The high symmetry of methane meant special symmetry considerations are required. Details of this are given in the papers.
Reference: [13YuTeBa], [14YuJe].
Model input files: YT10to10 spectroscopic model.
Spectroscopic Model MM
The model is described in [24YuOwTe].
KEO: Non-exact Taylor expansion around the equilibrium structure in terms of the valence (curvilinear) coordinates using the automatic differentiation (AD) technique [15YaYu] up to 6th order.
Coordinates: The choice of the valence coordinates is the same as used in 10to10, type R-SYM.
Frame: Eckart.
PEF: the same type general from the module pot_ch4.f90. A new ab initio PEF was refined to experimentally derived MARVEL energies of methane.
Potential expansion order: 8
Polyad scheme: 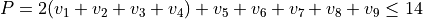 with caveats, see paper.
with caveats, see paper.
DMF: A new accurate ab initio DMS of the QZ quality.
Model input files: MM spectroscopic model.
Hydrogen peroxide, H2O2
The model (APTY) is described in [15AlOvYu], [16AlPoOv].
KEO: non-exact (linearised), expanded around a non-rigid reference configuration constructed to follow the torsion motion with all other valence coordinates fixed to their equilibrium values.
Symmetry: 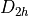 (EM).
(EM).
Frame: Eckart-Saywitz conditions with the x-axis in the plane bisecting the HOOH book-angle. The integration range for the torsional coordinate is extended to 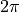 in order to efficiently separate the torsional and rotational degrees of freedom.
in order to efficiently separate the torsional and rotational degrees of freedom.
Coordinates: type (transform) r-alpha-tau. These are linearised except the torsional mode, based on the following valence-type coordinates,
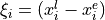 where
where 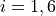 are
are 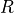 , , ,
, , , 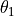 ,
, 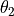 and .
and .
Molecular type (MOLTYPE): ABCD. This means that the coordinates, their symmetry properties and frame are defined in the module mol-ABCD.f90.
Coordinate to expand kinetic energy: , linearised coordinates
Coordinates to expand Potential energy: 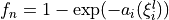
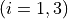 for stretches and
for stretches and
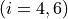 for bending coordinates.
for bending coordinates.
Potential linearised expansion order: 8
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
PEF: One of the integrated functional forms into the main, default TROVE compilation, type POTEN_H2O2_KOPUT_UNIQUE.
POT_TYPE POTEN_H2O2_KOPUT_UNIQUE
The PEF used was obtained by refining an ab initio CCSD(T)-F12b/aug-cc-pVNZ PES of HOOH to experimental ro-vibrational energies of the main isotopologue of HOOH , for 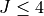 .
.
Basis set: Constructed using the polyad scheme: 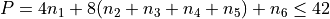 .
.
DMF type (DMS_TYPE): HOOH_MB. This dipole moment surface was computed using CCSD(T)-F12b/aug-cc-pV(T+d)Z and fitted to a polynomial.
Results: Linelist complete up to 6000 cm-1. Extended linelist up to 8000 cm-1 with reduced completeness at high temperatures.
Note
The coordinate for this molecule adds complications to expansion of dipole, etc. In order to guarantee a smooth torsional behaviour of all expansion terms of PEF and DMF, the iron-out feature was used. The iron-out card is placed anywhere of the main body of the input file (step 1) outside of any sections.
See papers for details.
Examples of the TROVE input file for the HOOH calculations using the APTY model can be found at APTY spectroscopic model.
Reference: [15AlOvYu], [16AlPoOv].
Formaldehyde, H2CO
Symmetry:
Coordinates: where are 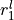 ,
, 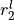 ,
, 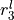 ,
, 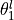 ,
, 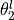 and .
and .
Coordinate to expand kinetic energy: 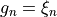 , linearised.
, linearised.
Coordinates type (transform): R-THETA-TAU
Coordinates to expand Potential energy: for stretches, 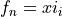 for bends.
for bends.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 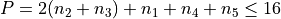 plus some additions, see paper.
plus some additions, see paper.
Potential energy function: CCSD(T)/aug-cc-pVQZ) ab initio energies fitted to polynomial expansion.
Refined using HITRAN data up to 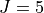 .
.
Dipole moment surface expansion: CCSD(T)/aug-cc-pVQZ ab initio dipole data fitted to polynomial expansion.
Results: Linelist for temperatures up to 1500 K for transitions up to 10,000 cm-1 and 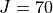 .
.
Reference: [15AlOvPo].
H2CS
TBP
Silane, SiH4
Symmetry: 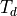
Coordinates: Linearised coordinates. As for methane.
Coordinate to expand kinetic energy: As for methane but with curvilinear coordinates.
Coordinates to expand Potential energy: As for methane.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 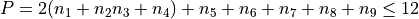 plus some additions, see paper.
plus some additions, see paper.
Potential energy function: CBS-F12 PES including extensive corrections, see paper. Fitted to polynomial expansion.
Refined using 1452 experimental energies up to 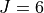 .
.
Dipole moment surface expansion: CCSD(T)/aug-cc-pVT(+d for Si)Z ab initio dipole data fitted to polynomial expansion.
Results: Linelist for temperatures up to 1200 K for transitions up to 5000 cm-1 and 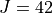 .
.
Reference: [17OwYuYa].
Methyl chloride, CH3Cl
Symmetry:
Coordinates: 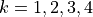 for vibrational coordinates,
for vibrational coordinates,
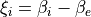 ,
, 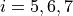 for bending coordinates,
for bending coordinates, 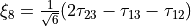 and
and 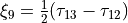 .
.
Coordinate to expand kinetic energy: , curvilinear coordinates used.
Coordinates to expand Potential energy: for stretches and
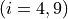 for bending coordinates.
for bending coordinates.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 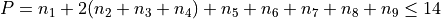 plus some additions, see paper.
plus some additions, see paper.
Potential energy function: CBS-F12 PES including extensive corrections, see paper. Fitted to polynomial form.
Dipole moment surface expansion: CCSD(T)/aug-cc-pVQZ(+d for Cl) level of theory. Fitted to polynomial form.
Results: Line list applicable up to 1200 K.
Note
Data for 35Cl and 37Cl isotopologues.
Reference: [15OwYuTa], [18OwYaTe] .
Ethylene, C2H4
Symmetry: 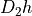
Coordinates: 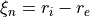
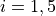 for stretches,
for stretches, 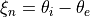
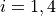 for bends,
for bends,
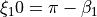 ,
, 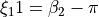 for two
for two 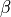 H-C-H ‘book type’ angles and
H-C-H ‘book type’ angles and
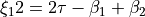 where is H-C-C-H dihedral angle.
where is H-C-C-H dihedral angle.
Coordinate to expand kinetic energy: . Curvilinear coordinates.
Coordinates to expand Potential energy: Morse coordinates for stretches, other coordinates expanded as 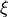 themselves.
themselves.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 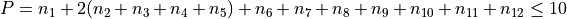 plus additions,
see paper.
plus additions,
see paper.
Potential energy function: ab initio PES calculated at CCSD(T)-F12b/cc-pVTZ-F12 level of theory. Fit to polynomial
form. Refined PES using HITRAN data for 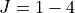 and other sources for vibrational band centres.
and other sources for vibrational band centres.
Dipole moment surface expansion: DMS calculated at CCSD(T)-F12b/aug-cc-pVTZ level of theory and fit to polynomial form with appropriate axis system.
Results: Line list for 0-7000 cm-1 up to 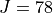 . Applicable up to 700 K.
. Applicable up to 700 K.
Note
Largest molecule in TROVE so far. Special techniques developed to cope with such a large molecule.
Reference: [18MaYaTe].
Phosphorus trifluoride, PF3
Symmetry:
Coordinates: 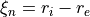
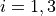 for stretching coordinates and
for stretching coordinates and 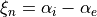 for bends.
for bends.
Coordinate to expand kinetic energy: . Linearised expansion.
Coordinates to expand Potential energy: Morse coordinates for stretches, bends expanded as themselves.
Primitive basis set: Numerov generated for all coordinates.
Kinetic energy expansion order: 6
Potential expansion order: 8
Polyad scheme: 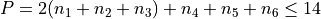 .
.
Potential energy function: ab initio PES calculated at CCSD(T)-F12b/cc-pVTZ-f12 level of theory fitted using polynomial expansion of symmetrized coordinates.
Dipole moment surface expansion: CCSD(T)/aug-cc-pVTZ ab initio dipole data fitted to polynomial expansion.
Results: Room temperature line list for up to 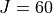 .
.
Note
The room temperature line list for this molecule is not complete but could be easily extended using the methods applied
to SO3 and C2H4.
Reference: [19MaChYa].