Dipole moment functions
TROVE provides a larger number of dipole moment functions (DMFs) for different molecules already implemented. Most of these PEFs are in modules pot_* contained in file pot_*.f90.
pot_xy2.f90
pot_xy3.f90
pot_zxy2.f90
pot_abcd.f90
pot_xy4.f90
pot_zxy3.f90
These are a part of the standard TROVE compilation set. Alternatively, a user-defined DMF can be included into the TROVE compilation as a generic ‘user-defined’ module pot_user.
Dipole (External) Block
The DMFs are defined in the TROVE input file using the Dipole block, which is just an alias for the external input structure. A typical Dipole input is as follows:
DIPOLE
dimension 3
NPARAM 264 0 0
compact
DMS_TYPE XY3_SYMMB
COEFF list
COORDS linear linear linear linear linear linear
Order 6 6 6
dstep 0.005
parameters
charge 0.00000000
nparamA 112.00000000
RhoE 90.00000000
RE14 1.01032000
beta 1.00000000
d0 4.56621083
f1a -9.36438932
f2a 32.96400671
...
end
For an example, see 14N-1H3__BYTe__TROVE__step1.inp where this DMF is used.
dimension(aliasesrank,dim): This is the dimension of the external field. An “external” is treated in TROVE as a vector of dimension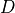 , which in the case of dipole can be up to
, which in the case of dipole can be up to 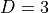 , but will depend on the implementation. This parameter is to help structure the input dipole parameters according to the dipole components, if necessary.
, but will depend on the implementation. This parameter is to help structure the input dipole parameters according to the dipole components, if necessary.NPARAMis used to specify the number of parameters to define the DMF and should contain input values.compact: is recently implemented card which switches to the “compact” format with no “fitting-indexes” column present.DMS_TYPE(TYPE) is the name of the DMF as implemented inpot_*.f90 fileand referenced inmlecules.f90.COEFFindicates if the DFM parameters are given as a list of parameter values (LIST) or values with the corresponding expansion powers (POWERS), see example below.COORDS: coordinate types used to re-expand the dipole field in terms of the internal TROVE coordinates.Order: The corresponding expansion order.dstep: finite difference step used in the re-expansion. The default value is 0.005 Ang.parameters: card indicating the section with the dipole parameters entries specific for the givenDMS_TYPE.
The dipole moment parameters are listed after the keyword parameters and terminated with the keyword END. The number of the entries should be equal exactly to the sum of NPARAM values.
For the COEFF list option, the meaning of the columns is as follows:
Label |
Value |
charge |
0.00000000 |
nparamA |
112.00000000 |
RhoE |
90.00000000 |
RE14 |
1.01032000 |
beta |
1.00000000 |
d0 |
4.56621083 |
f1a |
-9.36438932 |
f2a |
32.96400671 |
The first column with the name of the parameters, which is for clearness only. This field is only used for printing purposes and otherwise nor-referenced in the code in any way. The second column contains the actual value of the given parameter. The input is directly associated with the corresponding implementation and therefore the order is important.
An alternative, legacy, format with no compact card assumes an additional column with the so-called “fitting-indexes” indicating if the parameter was varied in the fitting to the ab initio data. Here is an example:
parameters
charge 0 0.00000000
nparamA 1 112.00000000
RhoE 0 90.00000000
RE14 0 1.01032000
beta 0 1.00000000
d0 4 4.56621083
f1a 8 -9.36438932
f2a 8 32.96400671
f3a 7 -80.82339377
....
where column 2 contains the “fitting-indexes”. These indexes are not used by TROVE. They are kept in order to simplify the interfacing between the ab initio fitting and TROVE, but can be always omitted with the help of the card compact.
Here is an example of the input format using individual expansion “powers”, COEFF powers (from CO2):
DIPOLE
rank 3
NPARAM 971 0 0
compact
TYPE DIPOLE_AMES1
COEFF powers (powers or list)
COORDS linear linear linear
Orders 16 16 16
threshold 1e-8
Parameters
re 0 0 0 0 1.15958d0
ae 0 0 0 0 180.00
d000 0 0 0 0 -0.4801402388843266D+00
d001 0 0 1 0 0.1203598337496481D+00
d002 0 0 2 0 -0.5662267278952241D-01
d003 0 0 3 0 -0.2529381009630170D-01
d004 0 0 4 0 -0.1271678002798687D+00
d005 0 0 5 0 0.3033049145401118D+01
d006 0 0 6 0 -0.1754036600894653D+02
.....
end
See the TROVE input CO2_bisect_xyz_step1.inp.
Assuming the DMF form as an expansion
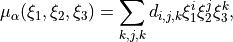
the input card has the following format
Label
Index
Value
d000
0
0
0
0
-0.4801402388843266D+00
d001
1
0
0
0
0.1203598337496481D+00
d002
0
1
0
0
-0.5662267278952241D-01
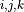
where
‘Labels’ are the parameter name, for printing purposes only;
,
,
are the ‘powers’ of an expansion term;
‘Index’ is a switch to indicate if the corresponding parameter was fitted or can be fitted, with no impact on any evaluations of the PEF values. It is not present in the
compactform.‘Values’ are the actual dipole parameters. For
powers, their order is not important.
In case the definition of DMF requires also structural parameters, such as equilibrium bond lengths 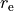 , equilibrium inter-bond angles
, equilibrium inter-bond angles 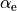 , in the
, in the COEFF Powers form these parameters should be listed exactly in the order expected by the implemented of the PEF (similar to the COEFF LIST form), but with dummy “powers” columns so that their ‘values’ appear in the right column, as in the example above, re and ae are two the equilibrium values and the three columns with 0 0 0 are given in order to parse their values using exactly column 6.
Implemented DMFs
XY2 type
See module pot_xy2.f90.
There are several PEFs available for this molecule type.
xy2_pq_coeff
This is a bisector-frame DMF, given by two components, 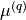 and
and 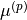 with the
with the 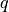 axis being the bisector. The following expansions in terms of the coordinate displacements
axis being the bisector. The following expansions in terms of the coordinate displacements 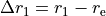 ,
, 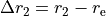 , and
, and 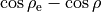 , where
, where 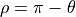 are used, with
are used, with 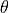 is the bond angle, and
is the bond angle, and 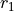 and
and 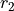 are the bond lengths:
are the bond lengths:
(1)![\begin{split}
\mu^{(q)} (\Delta r_1, \Delta r_2, \Delta \alpha ) &= \sin\alpha \left[ \mu_0^{(q)}(\alpha) + \sum_{j} \mu_{j}^{(q)}(\alpha) \Delta r_j + \sum_{j\le k} \mu_{jk}^{(q)}(\alpha) \Delta r_j \Delta r_k \right. \\
& \left . + \sum_{j\le k \le m} \mu_{jkm}^{(q)}(\alpha) \Delta r_j \Delta r_k \Delta r_m + \sum_{j\le k \le m \le n} \mu_{jkmn}^{(q)}(\alpha) \Delta r_j \Delta r_k \Delta r_m \Delta r_n + \ldots \right], \\
\mu^{(p)} (\Delta r_1, \Delta r_2, \Delta \alpha ) &= \mu_0^{(p)}(\alpha) + \sum_{j}^{(p)} \mu_{j}^{(p)} (\alpha) \Delta r_j + \sum_{j\le k} \mu_{jk}^{(p)}(\alpha) \Delta r_j \Delta r_k \\
& + \sum_{j\le k \le m} \mu_{jkm}^{(p)}(\alpha) \Delta r_j \Delta r_k \Delta r_m + \sum_{j\le k \le m \le n} \mu_{jkmn}^{(p)}(\alpha) \Delta r_j \Delta r_k \Delta r_m \Delta r_n + \ldots ,
\end{split}](_images/math/42fb58b4c2dea305da8ffedf5b61faf16044a177.png)
where all indices 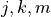 , and
, and 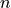 assume the values 1 or 2,
assume the values 1 or 2,
(2)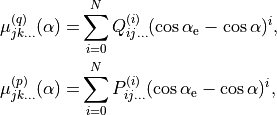
and the 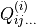 and
and 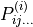 are molecular dipole parameters. The expansion coefficients in Eqs. (2) are subject to the conditions that the functions are unchanged under the interchange of the identical protons, whereas the function is antisymmetric under this operation. There are 72 and 99 paramters and , respectively. An example of
are molecular dipole parameters. The expansion coefficients in Eqs. (2) are subject to the conditions that the functions are unchanged under the interchange of the identical protons, whereas the function is antisymmetric under this operation. There are 72 and 99 paramters and , respectively. An example of xy2_pq_coeff is illustrated above and can be foound in H2S_EKE_basic-functions_step1.inp.
The implementation can be found in subroutine MLdms2pqr_xy2 from the module pot_xy2.f90. The transformation between the TROVE frame and the frame of the specifc dipole of the XY2 is perfomed in the subroutine MLloc2pqr_xy2, e.g.:
!
select case(trim(molec%frame))
!
case('R-RHO-Z','R-RHO-Z-M2-M3','R-RHO-Z-M2-M3-BISECT','BISECT-Z')
!
a0(2, 1) = -r(1) * cos(alpha_2)
a0(2, 3) = -r(1) * sin(alpha_2)
!
a0(3, 1) = -r(2) * cos(alpha_2)
a0(3, 3) = r(2) * sin(alpha_2)
case ...
XY2_PQ_LINEAR
This is similar to xy2_pq_coeff, but with the bending expansion in Eq. :eq:` e-muQ-exp` in terms of the displacement  :
:
(3)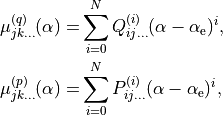
DIPOLE_AMES1
This DMF is of the AMES1 type represented using the point-charge molecular bond frame [14HuScLe] given by projections on the molecular bond vectors 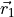 and
and 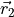 :
:
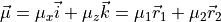
where 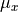 and
and 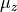 are the TROVE frame vectors and
are the TROVE frame vectors and 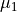 and
and 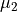 are the ab initio dipoles in the molecular bond frame (the
are the ab initio dipoles in the molecular bond frame (the 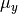 component is always zero). The two point-charge dipole moment components and are represented in terms of the vibrational coordinates as
component is always zero). The two point-charge dipole moment components and are represented in terms of the vibrational coordinates as
(4)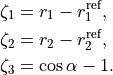
with the following analytic Taylor-type expansions used (see e.g. [14HuScLe]):
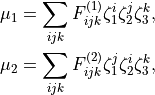
As an example can be found of a system where this form was used, see CO2_bisect_xyz_step1.inp.
DIPOLE_SO2_AMES1
This form is essentially the same as DIPOLE_AMES1 but some specific characteristic used for the SO2 molecule in [14HuScLe].
XY2_C3_SCHROEDER
This DMF is based on the DMF form reported by Schroeder et al. [16ScSe] for C3. This DMF is in the Ecakrt frmame expressed in terms of two in-plane components, 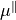 and
and 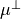 , as Taylor expansions around the equilibrium geometry:
, as Taylor expansions around the equilibrium geometry:
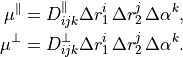
Since TROVE’s frame is usually different from the DMF frame (e.g. bisector) in the ro-vibrational calculations, this dipole moments functions needs to be rotated. This is done using the rotation angle 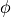 from an equilibrium bysector frame
from an equilibrium bysector frame 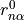 to the instantaneous frame
to the instantaneous frame 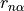 (
(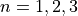 and
and 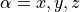 ) in the in the
) in the in the 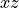 plane as given by
plane as given by
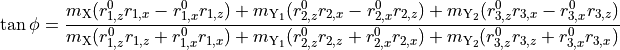
DIPOLE_PQR_XYZ_Z-FRAME
This is frame used to represent DMF of XYZ non-symmetri molecules with the  (
(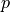 ) axis along the vecror and other two axes defined using the following conditions:
) axis along the vecror and other two axes defined using the following conditions:
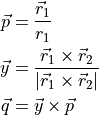
The corrsponding components and are expanded using the same form as in Eq. (1) but with no constraints on the permutations of the atoms.
DIPOLE_PQR_XYZ_Z-FRAME_SINRHO
The same as DIPOLE_PQR_XYZ_Z-FRAME but with 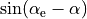 as an expansion variable in Eq. (1) instead of
as an expansion variable in Eq. (1) instead of  :
:
(5)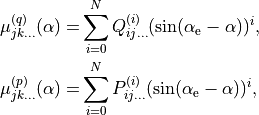
When 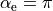 (linear molecules),
(linear molecules), 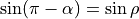 , which explanes the suffix
, which explanes the suffix _sinrho in the name of thi DMF, wich is aimed at linear molecules.
DIPOLE_PQR_XYZ
This a bisector dipole frame for the XYZ type molecules. It is defined by
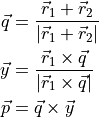
The exapnsion of the dipole moment components in terms of , and  as in Eq. (3). See
as in Eq. (3). See 7Li-16O-1H__OYT7__TROVE.model for an example of a TROVE input.
DIPOLE_PQR_XYZ_Z-BOND
Subroutine: MLdms2pqr_xyz_z_bond.
This is a generalisation of DIPOLE_PQR_XYZ_Z-FRAME, which does not make any assumtion on the frame of the original dipole, only on its expansion form given as in Eq. (3). The role of DIPOLE_PQR_XYZ_Z-BOND is to transform it to the TROVE frame, which in this case is the with the $z$ axis oriented along the bond :
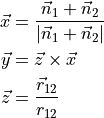
DIPOLE_PQR_XYZ_BISECTING
Subroutine: MLdms2pqr_xyz_bisecting.
This is a generalisation of DIPOLE_PQR_XYZ, which does not make any assumtion on the frame of the original dipole, only on its expansion form given as in Eq. (3). The role of DIPOLE_PQR_XYZ_BISECTING is to transform it to the TROVE frame, which in this case is the with the $x$ axis oriented along the bisector:
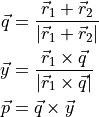
DIPOLE_AMES1_XYZ
This form is a modification of DIPOLE_AMES1 for non-symmetric molecules.
As an example can be found of a system where this form was used, see 16O-12C-32S__OYT8__TROVE.model as well in OYT8 spectroscopic model, where it was used to compute an ExoMol line list for OCS [24OwYuTe].
XY2_SCHROEDER_XYZ_ECKART
This is an XYZ version of the XY2_C3_SCHROEDER type.
DIPOLE_H2O_LPT2011
DMF from [11LoTePo]. It is included into subroutine MLdipole_h2o_lpt2011 in prop_xy2.f90.
DIPOLE_PQR_XYZ_Z-BOND
DIPOLE_PQR_XYZ_BISECTING`
DIPOLE_BISECT_S1S2T_XYZ
XY2_QMOM_SYM
XY2_ALPHA_SYM
XY2_QMOM_BISECT_FRAME
TEST_XY2_QMOM_BISECT_FRAME
XY2_SR-BISECT-NONLIN
TEST_XY2_SR-BISECT-NONLIN
XY3
See module pot_xy3.f90.
XY3_MB
This DMF is implemented as a subroutine MLdms2xyz_xy3_mb and was reported in [06YuCaTh]. This form was used to produce a line list for PH3 in [13SoYuTe]. An input example is in 31P-1H3__SAlTY__TROVE.model and can be also found at https://exomol.com/models/PH3/31P-1H3/SAlTY/.
Consider the 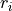 as the instantaneous value of the distance between X and Y
as the instantaneous value of the distance between X and Y , where Y is the nucleus labeled (= 1, 2, or 3);
, where Y is the nucleus labeled (= 1, 2, or 3); 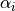 denotes the bond angle
denotes the bond angle 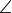 (Y
(Y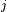 XY
XY ) where
) where 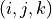 is a permutation of the numbers (1,2,3).
is a permutation of the numbers (1,2,3).
We utilize the so-called Molecular-Bond (MB) representation to describe the and 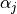 dependence of the electronically averaged
dipole moment vector
dependence of the electronically averaged
dipole moment vector 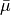 for XY3. In the MB representation it is given by
for XY3. In the MB representation it is given by
(6)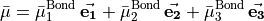
where the three functions 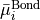 ,
,  1, 2, 3, depend on the vibrational coordinates, and
1, 2, 3, depend on the vibrational coordinates, and 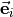 is the unit vector along bond ,
is the unit vector along bond ,
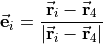
with 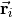 ( 1, 2, 3) as the position vector of proton and
( 1, 2, 3) as the position vector of proton and 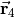 as the position vector of X. The representation of in Eq. (6) is “body-fixed” in the sense that it relates the dipole moment vector directly to the instantaneous positions of the nuclei (i.e., to the vectors ).
as the position vector of X. The representation of in Eq. (6) is “body-fixed” in the sense that it relates the dipole moment vector directly to the instantaneous positions of the nuclei (i.e., to the vectors ).
Following [06YuCaTh], we express the three functions , 1, 2, 3, as
(7)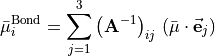
where 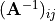 is an element of the non-orthogonal
is an element of the non-orthogonal 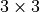 matrix
matrix 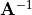 obtained as the inversefootnote%
{When the molecule is planar, the determinant
obtained as the inversefootnote%
{When the molecule is planar, the determinant  0 and
0 and  cannot be inverted. In this case we set
cannot be inverted. In this case we set  0 in Eq. (6) and express in terms of
0 in Eq. (6) and express in terms of 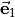 and
and 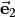 only, i.e., we determine
only, i.e., we determine 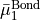 and
and 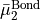 in terms of
in terms of 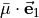 and
and 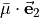 .} of
.} of
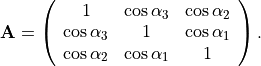
For symmetry reasons, all three projections can be expressed in terms of a single function  :
:
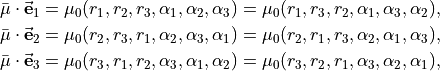
and this function is chosen as an expansion
(8)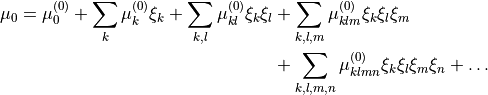
in the variables
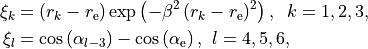
where and are the equilibrium values of the bond lengths and bond angles, respectively. We include the factor 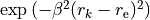 in order to keep the expansion from diverging at large
in order to keep the expansion from diverging at large 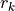 .
.
The expansion coefficients 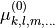 are pairwise equal. We have
are pairwise equal. We have
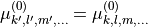
when the indices 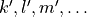 are obtained from
are obtained from 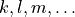 by replacing all indices 2 by 3, all indices 3 by 2, all indices 5 by 6, and all indices 6 by 5.
by replacing all indices 2 by 3, all indices 3 by 2, all indices 5 by 6, and all indices 6 by 5.
XY3_SYMMB
This form is a non-rigid analogy of XY3_MB allowing the pyrimidal molecule XY3 to go through the planar configuration and was introduced in [09YuBaYa]. It is implemented in subroutine MLdms2xyz_xy3_symmb in pot_xy3.f90. This form has been used in several studies of non-rigid pyrimidal molecules including NH3, CH3, OH3+.
A disadvantage of the XY3_MB representation is the ambiguity at and near planar geometries when the three vectors become linearly dependent, or nearly linearly dependent, and singularities appear in the determination of the functions. This is overcome by reformulating the functions in terms of symmetry-adapted combinations of the MB projections 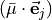
![\begin{split}
\mu_{A''_1}^{\rm SMB}&= \left( \bar{\bf \mu} \cdot \vec{\bf e}_{\rm N} \right) \\
\mu_{E'_a}^{\rm SMB} &= \frac{1}{\sqrt{6}} \left[ 2 \left( \bar{\bf \mu} \cdot \vec{\bf e}_1 \right) - \left( \bar{\bf \mu} \cdot \vec{\bf e}_2 \right) - \left( \bar{\bf \mu} \cdot \vec{\bf e}_3 \right) \right] \\
\mu_{E'_b}^{\rm SMB} &= \frac{1}{\sqrt{2}} \left[ \left( \bar{\bf \mu} \cdot \vec{\bf e}_2 \right) - \left( \bar{\bf \mu} \cdot \vec{\bf e}_3 \right) \right],
\end{split}](_images/math/a4118f23a117d0827548aa0e598d8ffe44b410b6.png)
where an additional reference MB-vector 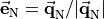 was introduced by means of the ‘trisector’
was introduced by means of the ‘trisector’
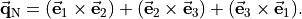
This symmetrized molecular bond representation is denoted as “SMB”. The subscripts of the 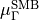 functions (
functions (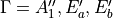 ) refer to irreducible representations of D3h(M);
) refer to irreducible representations of D3h(M); 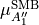 has
has 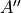 symmetry in D3h(M), and
symmetry in D3h(M), and 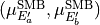 transform as the
transform as the  irreducible representation. The symmetrized vectors
irreducible representation. The symmetrized vectors
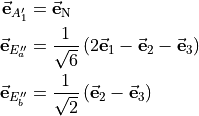
have  and
and 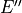 symmetry in the same manner.
symmetry in the same manner.
The dipole moment vector vanishes at symmetric, planar configurations of D3h geometrical symmetry. Also, the 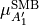 component is antisymmetric under the inversion operation
component is antisymmetric under the inversion operation 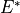 and vanishes at planarity, which leaves only two independent components of at planarity.
and vanishes at planarity, which leaves only two independent components of at planarity.
The advantage of having a DMS representation in terms of the projections 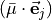 is that it is “body-fixed”. It relates the dipole moment vector directly to the instantaneous positions of the nuclei (i.e., to the vectors ). These projections are well suited to being represented as analytical functions of the vibrational coordinates. For intensity simulations, however, we require the Cartesian components
is that it is “body-fixed”. It relates the dipole moment vector directly to the instantaneous positions of the nuclei (i.e., to the vectors ). These projections are well suited to being represented as analytical functions of the vibrational coordinates. For intensity simulations, however, we require the Cartesian components 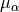 , of the dipole moment along the molecule-fixed
, of the dipole moment along the molecule-fixed 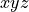 axes. These are obtained by inverting the linear equations
axes. These are obtained by inverting the linear equations
(9)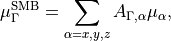
where 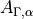 , is the -coordinate () of the vector
, is the -coordinate () of the vector 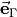 (
(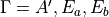 ). When the molecule is planar, is zero, as is the corresponding right-hand side in Eq. (9). Thus, at planar configurations the system of linear equations in contains two non-trivial equations only. At near-planar configurations is not exactly zero and cannot be neglected, and so Eq. (9) becomes near-linear-dependent. The symmetry-adapted representation of
). When the molecule is planar, is zero, as is the corresponding right-hand side in Eq. (9). Thus, at planar configurations the system of linear equations in contains two non-trivial equations only. At near-planar configurations is not exactly zero and cannot be neglected, and so Eq. (9) becomes near-linear-dependent. The symmetry-adapted representation of 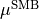 appears to be well defined even for these geometries.
appears to be well defined even for these geometries.
The functions (henceforth referred to as 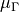 ) are now represented as
expansions:
) are now represented as
expansions:
![\begin{split}
\mu_{A''_1} & = \cos\rho \left[ \mu_{0}^{(A''_1)} + \sum_{k} \mu_{k}^{(A''_1)} \xi_k + \sum_{k,l} \mu_{k,l}^{(A''_1)} \xi_k \xi_l + \sum_{k,l,m} \mu_{k,l,m}^{(A''_1)} \xi_k \xi_l \xi_m + \cdots \right] \\
\mu_{E'_a} & = \mu_{0}^{(E'_a)} + \sum_{k} \mu_{k}^{(E'_a)} \xi_k + \sum_{k,l} \mu_{k,l}^{(E'_a)} \xi_k \xi_l + \sum_{k,l,m} \mu_{k,l,m}^{(E'_a)} \xi_k \xi_l \xi_m + \cdots \\
\mu_{E'_b} = & \mu_{0}^{(E'_b)} + \sum_{k} \mu_{k}^{(E'_b)} \xi_k + \sum_{k,l} \mu_{k,l}^{(E'_b)} \xi_k \xi_l + \sum_{k,l,m} \mu_{k,l,m}^{(E'_b)} \xi_k \xi_l \xi_m + \cdots
\end{split}](_images/math/5ff9ef8310a87bc7f6ff0c862853ac51cc506c34.png)
in terms of the variables
![\begin{split}
\xi_k &= (r_k - r_{\rm e}) \exp \left[ -\beta \, (r_k-r_{\rm e})^2 \right], \;\; k=1,2,3, \\
\xi_{4}&= \frac{1}{\sqrt{6}} \left( 2 \alpha_{1} - \alpha_{2} - \alpha_{3} \right)\hbox{,} \\
\xi_{5}&= \frac{1}{\sqrt{2}} \left( \alpha_{2} - \alpha_{3} \right)\hbox{,} \\
\xi_{6} &= \sin\rho_{\rm e}-\sin\rho \hbox{,}
\end{split}](_images/math/50bd2372d35909a41b1e29d046472dd1b3b06579.png)
Here
![\sin {\rho} \, = \, \frac{2}{\sqrt{3}} \, \sin [ (\alpha_1 + \alpha_2 + \alpha_3) /6],](_images/math/88da169a4ccabd6bf6a9d5e95a35e13492497f78.png)
and 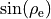 is the equilibrium value of
is the equilibrium value of 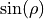 . The factor
. The factor 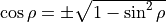 ensures that the dipole moment function
ensures that the dipole moment function 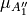 changes sign when
changes sign when 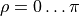 is changed to
is changed to 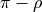 . As in the rigid MB case, the factor
. As in the rigid MB case, the factor ![\exp \left[ -\beta (r_k-r_{\rm e})^2 \right]](_images/math/4ebcc659b897f8b44b3e07489009806306fb5bb7.png) is used in order to keep the expansion from diverging at large .
is used in order to keep the expansion from diverging at large .
Chain type ABCD-type molecules
HOOH_MB
Subroutine: MLdms_hooh_MB.
The DMF frame is defined with the 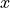 axis as a bisector between two planes, H1O1O2 and O1O2H1 and axis along the O1O2 bond. Thus, the unit vector is defined as a normalised vector
axis as a bisector between two planes, H1O1O2 and O1O2H1 and axis along the O1O2 bond. Thus, the unit vector is defined as a normalised vector 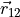 , the unit vector is defined as a bisector between the normal to the planes 3-1-2 and 1-2-3 and the unit vector is defined via the rand-hand rule:
, the unit vector is defined as a bisector between the normal to the planes 3-1-2 and 1-2-3 and the unit vector is defined via the rand-hand rule:
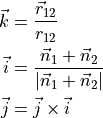
where the plane normals are defined as follows:
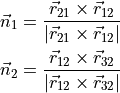
Here, the coordinate vectors 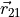 ,
, 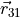 and
and 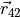 as well as
as well as 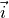 ,
, 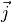 and
and 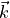 are given in the representation of the instantaneous TROVE frame . With this definition, the matrix constructed from these unit vecrtors , and forms the rotation (unitary transformation) of the dipole vector between its original DMF frame and the instantaneous TROVE frame :
are given in the representation of the instantaneous TROVE frame . With this definition, the matrix constructed from these unit vecrtors , and forms the rotation (unitary transformation) of the dipole vector between its original DMF frame and the instantaneous TROVE frame :
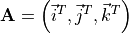
and
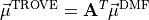
The 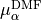 are expressed as Taylor-type expansion:
are expressed as Taylor-type expansion:
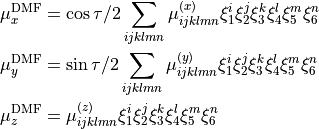
where
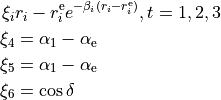
The expansion paramters are the subjetc to the permutation constraint:
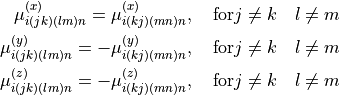
This DMF form was used to produce the APTY line list for HOOH [15AlOvYu]. The TROVE input file describing this spectroscopic model can be found in APTY spectroscopic model. For this mode, the Dipole block is given by
Dipole
dimension 3
NPARAM 136 96 98
DMS_TYPE HOOH_MB
COEFF powers
COORDS linear
Order 6 6 6
parameters
re1 0 0 0 0 0 0 0 1.45538654
re2 0 0 0 0 0 0 0 0.96257063
alphae 0 0 0 0 0 0 0 101.08307909
beta1 0 0 0 0 0 0 0 1.00000000
beta2 0 0 0 0 0 0 0 1.00000000
xxxxx 0 0 0 0 0 0 0 0.00000000
mu000000 0 0 0 0 0 0 7 3.11323258
mu000001 0 0 0 0 0 1 6 -0.05725254
mu000002 0 0 0 0 0 2 5 -0.00374176
mu000003 0 0 0 0 0 3 4 -0.0048
....
where dimension is 3.
HPPH_MB
Subroutine: MLdms_hpph_MB
See ./input/39K-16O-1H__OYT4_model_TROVE.inp
HCCH_MBHCCH_DMS_7DHCCH_DMS_7D_7ORDERHCCH_DMS_7D_7ORDER_LINEARHCCH_DMS_7D_LOCALHCCH_ALPHA_ISO_7D_LINEAR
ZXY2
ZXY2_SYMADAP
ZXY3
ZXY3_SYM
C2H4
DIPOLE_C2H4_4M
Properties
XY2_SR-BISECTXY2_SS_DIPOLE_YY
XY2_G-BISECTXY2_G-ROT-ELECXY2_G-COR-ELECXY2_G-TENS-RANK3XY2_G-TENS-NUCXY3_NSS_MBCOORDINATES